In my experience, one of the most often used claims for the alleged strength of evolutionary theory is the ability of researchers to construct phylogenetic trees which are said to represent the evolutionary development of major taxonomic groups of life. The main idea conveyed is that such seemingly congruent diagrams would not be possible to construct if Common Descent were false... that is to say, If Common Descent were false, researchers would run into major irreconcilable contradictions while attempting to plot out common ancestry relationships between various major types of life.
My contention here is that the above claim is wrong, and that Phylogeny is, for the most part, unfalsifiable. Instead, what Phylogenetic tree construction appears to be is a Metaphysical statistics program. What I mean is that such systematic constructions proceed only within the prior assumption that Common Descent is true. There are rigorously complex systematic and statistical models for attempting to resolve the 'most-likely' pattern of Common Descent (if it is true), but the model itself lacks any substantial criteria for falsifying the evolutionary assumption it is based on.
The reason I contend this is because of the amount of evolutionary rescue-devices available to reconcile nearly any amount of incongruent or contradictory data.
I will attempt to demonstrate this with the following examples:
First, to show an example of major contradiction in the data... Compare the Human/Mouse/Dog relationship shown above with this study published a year later....
So in a full-genome study we see "overwhelming support" for Primates/Carnivore (Human-Dog) clade, to the exclusion of rodents. But the first study using retroposon insertion sites shows a Primate/Rodent clade with the exclusion of Carnivores(Dogs). This demonstrates that major contradictions are not a problem for phylogeny.
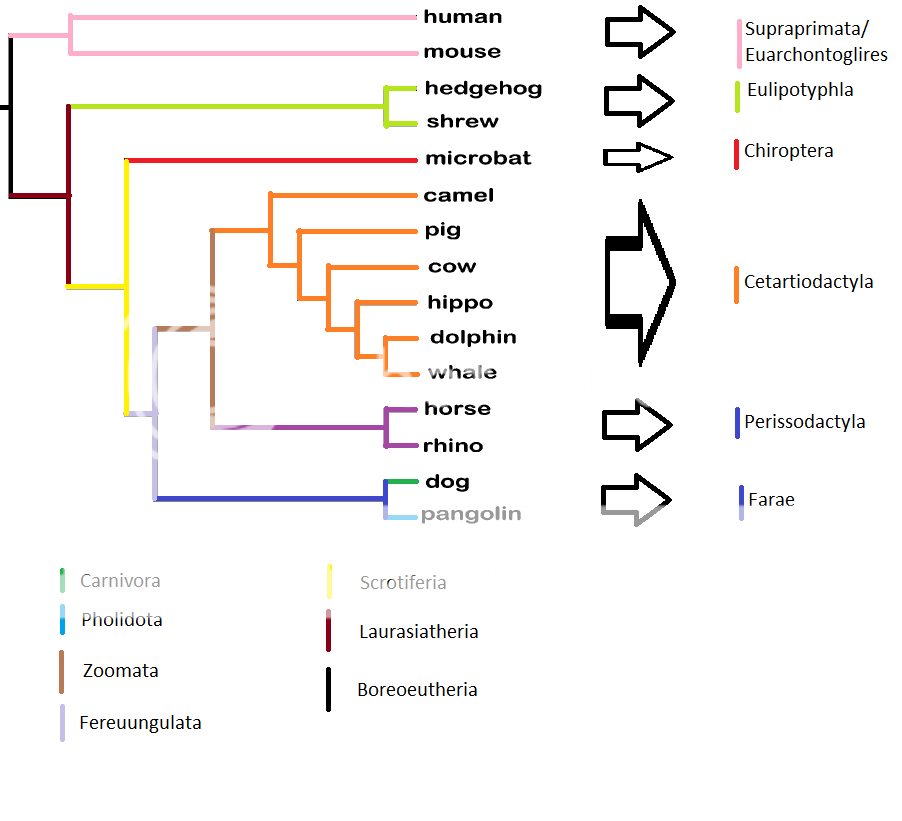
Now, returning to the original Pegasoferae paper, we will highlight a rescue device known as "incomplete lineage sorting" that can potentially be used to explain away large amounts of discordant data.
Here is the explanation for the retroposon presence in the Cetartiodactyla group:

Looking at the tree image again: The researchers found the ERV: INT283 in Perissodactyla, Carnivora, and Certartiodactyla. However Certartiodactyla lacks the four ERVs(165,265,382,391) that unite Pegasoferae. They don't want to propose that INT283 is a homoplasy, (or independently acquired in one group), so the researchers simply conclude that an "incomplete sorting" event took place somewhere between the common ancestor of Pegasoferae and Scrotifera, whereby the lineage representing Certiodactyla avoided gene pools containing the "Pegasoferae" ERV's, but later recombined with gene pools containing the ERV INT283.
Using this methodology, it seems nearly any pattern of retroposon integration could be explained. As a quick experiment, I took the phylogenetic tree from the same study and simulated a new data point that forced me to completely rearrange part of the tree, though I only invoked the same devices as the original paper.
Here's a crude modification of the original diagram with my simulated data point: the presence of a retroposon integration marker: INT138 located in the order Glires (comprised of Rodentia, Lagamoprha). From this I was able to "resolve" a new clade named "Gligasoferae". I included a mock abstract to go with the simulated data.
The point of this was to give a simple example of how easily different data sets can be explained with evolutionary rescue devices such as incomplete lineage sorting.
Evolutionists may counter that "incomplete lineage sorting" is observed in living populations and is an inevitable product of population genetics. But this is not the point. The point is that the evolutionary model is predicting a scenario of unfalsifiability, where discordant data always has an explanation. Regardless of whether or not it is a possible explanation, the model becomes largely removed from falsifiable criteria. The discordant gene trees might be a product of "incomplete lineage sorting", or it might represent true contradictions to phylogeny because Evolution didn't happen.
The latter hypothesis is metaphysically or philosophically unacceptable to evolutionists, so they will simply explain any amount of gene tree discordance as incomplete lineage sorting, or other evolutionary rescue devices.
I believe this is a reoccurring pattern for most of the alleged strengths of the theory of Evolution. Its robustness is an illusion....
For another example: Here's another ERV-based construction attempting to show the phylogeny of the major orders of placental mammals. They found that retroposon integration sites significantly contradicted any single evolutionary tree, which will be more apparent in the images below.
Isn't it interesting how severely conflicting data just becomes more confirming evidence for Evolution?
This last image (figures A) says it all. Look at the liberty in which the Afrotheria lineage is permitted to skip over the first set of insertions assigned to Boreotheria, but then combines with Boreotheria's second set insertions, finally combining with Xenartha insertions, (Xenartha containing insertions from Boreotheria's first set, but not second)
Here the researchers use the same rescue devices to resolve blatantly contradictory data on a larger scale.
This type of behavior supports my suspicions that Evolutionary Phylogenetics is a metaphysical or philosophical statistics/systematics program, that is largely unfalsifiable. While robust comparative methodology is applied within the model, the model itself does not have a falsifiable scientific basis but a metaphysical one. That is, its practitioners hold a metaphysical, philosophical, or religious conviction that Evolution / Common Descent is true.
I believe this metaphysical conviction to Evolution stems from the root desire to explain existence while denying the obvious: that the Creation had a Creator. But I suppose that is another subject altogether.
My contention here is that the above claim is wrong, and that Phylogeny is, for the most part, unfalsifiable. Instead, what Phylogenetic tree construction appears to be is a Metaphysical statistics program. What I mean is that such systematic constructions proceed only within the prior assumption that Common Descent is true. There are rigorously complex systematic and statistical models for attempting to resolve the 'most-likely' pattern of Common Descent (if it is true), but the model itself lacks any substantial criteria for falsifying the evolutionary assumption it is based on.
The reason I contend this is because of the amount of evolutionary rescue-devices available to reconcile nearly any amount of incongruent or contradictory data.
I will attempt to demonstrate this with the following examples:
Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions
Nishihara et al. 2006
....We propose a superordinal name “Pegasoferae” for this clade of Chiroptera + Perissodactyla + Carnivora + Pholidota. The presence of a single incongruent L1 locus generates a tree in which the group of Carnivora + Perissodactyla associates with Cetartiodactyla but not with Chiroptera. This result suggests that incomplete lineage sorting of an ancestral dimorphism occurred with regard to the presence or absence of retroposon alleles in a common ancestor of Scrotifera (Pegasoferae + Cetartiodactyla), which was followed by rapid divergence into the extant orders over an evolutionarily short period.
http://www.pnas.org/content/103/26/9929.full#B2
First, to show an example of major contradiction in the data... Compare the Human/Mouse/Dog relationship shown above with this study published a year later....
A Phylogenomic Study of Human, Dog, and Mouse
Cannarozzi et al. 2007
In recent years the phylogenetic relationship of mammalian orders has been addressed in a number of molecular studies. These analyses have frequently yielded inconsistent results with respect to some basal ordinal relationships. For example, the relative placement of primates, rodents, and carnivores has differed in various studies. Here, we attempt to resolve this phylogenetic problem by using data from completely sequenced nuclear genomes to base the analyses on the largest possible amount of data. To minimize the risk of reconstruction artifacts, the trees were reconstructed under different criteria—distance, parsimony, and likelihood. For the distance trees, distance metrics that measure independent phenomena (amino acid replacement, synonymous substitution, and gene reordering) were used, as it is highly improbable that all of the trees would be affected the same way by any reconstruction artifact. In contradiction to the currently favored classification, our results based on full-genome analysis of the phylogenetic relationship between human, dog, and mouse yielded overwhelming support for a primate–carnivore clade with the exclusion of rodents.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1761043/
So in a full-genome study we see "overwhelming support" for Primates/Carnivore (Human-Dog) clade, to the exclusion of rodents. But the first study using retroposon insertion sites shows a Primate/Rodent clade with the exclusion of Carnivores(Dogs). This demonstrates that major contradictions are not a problem for phylogeny.
Now, returning to the original Pegasoferae paper, we will highlight a rescue device known as "incomplete lineage sorting" that can potentially be used to explain away large amounts of discordant data.
Here is the explanation for the retroposon presence in the Cetartiodactyla group:
Characterization of an Inconsistent Locus.
In addition to the four loci that support the Pegasoferae clade, we isolated one L1 locus (INT283) that indicates the monophyly of Carnivora, Perissodactyla, and Cetartiodactyla, supporting Fereuungulata. The probability of homoplasy can be considered extremely low for L1 insertions, and the same target-site duplications (that are produced in the process of integration) were clearly observed for both patterns of the insertions (Figs. 3 B and 4F). Thus, this inconsistency cannot be explained by homoplasy, and both of the incongruent trees are convincing gene trees (40). If clear homoplasy-free genetic markers show incongruent trees, it can be interpreted that lineage sorting of polymorphism in a common ancestor of the species was incomplete. Namely, the L1 insertion at locus INT283 occurred in the genome of a common ancestor of Scrotifera, and the ancestral dimorphism of alleles containing or lacking L1 had been retained in the population during the divergence of at least three lineages such as Cetartiodactyla, Chiroptera, and a group of Carnivora + Perissodactyla, followed by random fixation of the alleles. Accordingly, the time period in which the divergences occurred is relatively shorter than the coalescence time of the ancestral population (22, 40). Waddell et al. (8) proposed a likelihood analysis to estimate significance for the lineage supported by retroposon method. According to the estimation, the clade supported by four loci is statistically significant (P = 0.025; ref. 8) despite the existence of one incongruent locus. Therefore, it seems likely that the monophyly of Carnivora, Perissodactyla, and Chiroptera reflect the true species’ relationships, and Fereuungulata (including Carnivora, Perissodactyla and Chiroptera) supported by one locus can no longer be considered a robust clade.
Looking at the tree image again: The researchers found the ERV: INT283 in Perissodactyla, Carnivora, and Certartiodactyla. However Certartiodactyla lacks the four ERVs(165,265,382,391) that unite Pegasoferae. They don't want to propose that INT283 is a homoplasy, (or independently acquired in one group), so the researchers simply conclude that an "incomplete sorting" event took place somewhere between the common ancestor of Pegasoferae and Scrotifera, whereby the lineage representing Certiodactyla avoided gene pools containing the "Pegasoferae" ERV's, but later recombined with gene pools containing the ERV INT283.
Using this methodology, it seems nearly any pattern of retroposon integration could be explained. As a quick experiment, I took the phylogenetic tree from the same study and simulated a new data point that forced me to completely rearrange part of the tree, though I only invoked the same devices as the original paper.
Here's a crude modification of the original diagram with my simulated data point: the presence of a retroposon integration marker: INT138 located in the order Glires (comprised of Rodentia, Lagamoprha). From this I was able to "resolve" a new clade named "Gligasoferae". I included a mock abstract to go with the simulated data.

Following the discovery of the presence of the enigmatic retroposon insertion (INT138), we recover the order Glires (Rodents/Rabbits), as belonging to a clade that is shared with Pegasoferae(Chrioptera, Perissodactyla, Carnivora). We name this newly resolved clade "Gligasoferae".
Since the probability of homoplasy of INT138 is highly unlikely, this clade is resolved by two occurrences of incomplete lineage sorting. (shown in blue labeled ILS1, and ILS2) ILS 1 occurs within common ancestors of the magnorder Boreoeutheria, whereby populations containing insertion sites "A" (labeled in green) were combined with an incompletely sorted polymorphism lacking fixation of insertion sites "B". ILS 2 shows the lineage representing order Glires shared a common ancestor with an incompletely sorted polymoprhism population in the common ancestor of the clade Scrotifera, which had inherited retroposon INT138 (shown in red), but lacked fixation of INT226, INT237.
The point of this was to give a simple example of how easily different data sets can be explained with evolutionary rescue devices such as incomplete lineage sorting.
Evolutionists may counter that "incomplete lineage sorting" is observed in living populations and is an inevitable product of population genetics. But this is not the point. The point is that the evolutionary model is predicting a scenario of unfalsifiability, where discordant data always has an explanation. Regardless of whether or not it is a possible explanation, the model becomes largely removed from falsifiable criteria. The discordant gene trees might be a product of "incomplete lineage sorting", or it might represent true contradictions to phylogeny because Evolution didn't happen.
The latter hypothesis is metaphysically or philosophically unacceptable to evolutionists, so they will simply explain any amount of gene tree discordance as incomplete lineage sorting, or other evolutionary rescue devices.
I believe this is a reoccurring pattern for most of the alleged strengths of the theory of Evolution. Its robustness is an illusion....
For another example: Here's another ERV-based construction attempting to show the phylogeny of the major orders of placental mammals. They found that retroposon integration sites significantly contradicted any single evolutionary tree, which will be more apparent in the images below.
Mosaic retroposon insertion patterns in placental mammals
Churakov 2009
...These findings provide significant support for a “soft” polytomy of the major mammalian clades. Ancestral successive hybridization events and/or incomplete lineage sorting associated with short speciation intervals are viable explanations for the mosaic retroposon insertion patterns of recent placental mammals and for the futile search for a clear root dichotomy.
.....We believe that the most parsimonious interpretation of the current data is that the ancestral placental populations were characterized by severe ancestral subdivisions and rejoinings, leading to a complex mosaic of phylogenetic relationships in recent species (Fig. 4). Effects of alternating divergence, hybridization, introgression, and incomplete lineage sorting might complicate our search for a clear dichotomy at the base of this tree and leave us with an indistinct, effective ‘soft’ polytomy, leading sometimes to one or the other solution depending on the size of the data set and the particular markers examined.

In the first scenario, the Eomaia-like ancestral population probably diverged into three distinct lineages of preAfrotheria, preXenarthra, and preBoreotheria. Limited gene flow and hybridization that occurred among these populations left behind mosaic signs of intermittent relationships that we identify about 125 million years later as a patchwork of Exafroplacentalia (I), Epitheria (II), and Atlantogenata (III) roots (Fig. 4A). This scenario requires temporary overlap of the three lineages or parts of their populations before final speciation occurred (Fig. 4B). A second scenario that could explain the results involves incomplete lineage sorting among the three populations (Fig. 4C). Because retroposon fixation requires possibly millions of years before being consistently represented in a population, alleles with and without certain elements surely coexisted temporarily, and, in the case of rapid speciation, were distributed randomly into different lineages. Such a process could contribute as well to the patchwork presentation of seemingly competing insertion patterns now present in recent species....
....The present example of apparently incongruent markers inherent in the early branching of placentals offers a seminal example of conflicting retroposon presence/absence patterns that, at the same time, shed new light on the decades-old, controversial scenarios of sequence-based phylogenies.

http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2675975/
Isn't it interesting how severely conflicting data just becomes more confirming evidence for Evolution?
This last image (figures A) says it all. Look at the liberty in which the Afrotheria lineage is permitted to skip over the first set of insertions assigned to Boreotheria, but then combines with Boreotheria's second set insertions, finally combining with Xenartha insertions, (Xenartha containing insertions from Boreotheria's first set, but not second)
Here the researchers use the same rescue devices to resolve blatantly contradictory data on a larger scale.
This type of behavior supports my suspicions that Evolutionary Phylogenetics is a metaphysical or philosophical statistics/systematics program, that is largely unfalsifiable. While robust comparative methodology is applied within the model, the model itself does not have a falsifiable scientific basis but a metaphysical one. That is, its practitioners hold a metaphysical, philosophical, or religious conviction that Evolution / Common Descent is true.
I believe this metaphysical conviction to Evolution stems from the root desire to explain existence while denying the obvious: that the Creation had a Creator. But I suppose that is another subject altogether.
")