Nesslig20
Active Member
I have come in contact with a creationist youtuber Standing For Truth (SfT) and he wants to have a talk with me and others in the near future, although I have yet to get a response from him to know exactly when he wants to talk. I have listened to him multiple conversations with people like Jackson Wheat, Crispr and Scientist Sam and I can summarise his tactic of discourse as such:
JAQing off (short for "Just-asking-questions")
It’s a way to phrase claims as questions in such a way that completely disregards the answers that could be given and is only intended to influence the views of spectators. For example, if an 9/11 truther asks questions about how you can explain their perceived evidence of a controlled demolition, it implies that IF you or someone else is unable to answer the question on the spot then they are seemingly winning the point automatically, hence it WAS evidence of a controlled demolition. It’s a clever way to shift the burden of proof. So SfT often asks questions with claims and assertion hidden within them. That's one thing to watch out for. SfT also employs changing topic constantly by asking new questions immediately after previous one is answers and once he is pinned into a corner (the opponent won't let him change the subject), he apparently begins to freak out and starts spouting a string of nonsense all at once just to flabbergast the opponent and distract them with this steaming pile of BS. A discussion with him would be quite some experience. And I believe that his tactic would work more effectively with people who aren’t prepared to answer his loaded/leading questions, so here I have prepared some answers/rebuttals to his questions/assertions.
One of his main talking points is new genetic information, in which he seems to define as “new functional DNA popping out of nothing”. Note the “out of nothing” caveat. It doesn’t matter if you have a completely new gene forming from non-coding DNA which previously didn’t do anything, it doesn’t count as new information since the template was already there. Of course, Evolution doesn’t entail nor require that things pop out of thing air. Evolution is more like tinkering. Rather than inventing new things it uses old stuff and modifies it to make the new. A good example of this is the GPCR (G-coupled Protein receptors) superfamily. They bind to molecules outside the cell and when that happens they activate an internal signal transduction pathway. It is an extremely fast way for cells to respond to external stimuli. The first GPCR got duplicated, and the duplicated version got duplicated in turn, and so forth, creating this protein super family.

Each duplicated version was slightly modified such that they would react to different molecules. This produced a wide variety of different GPCRs with completely different functions in many systems. There are those that bind to hormones, regulate the immune system and inflammation, some are involved in our sense of smell and taste because they bind to flavour and odour molecules. Some GPCRs even got modified such that they could react to light and are now the opsins in the rod cells of our eyes. It's rather odd that creationists are asking us to show new information, i.e. show how DNA can pop into existence from nothing.
That’s the creationists view, God making things from nothing. So they should be asking this question to themselves, not us.
It also shows that they have this conception that these biological systems just HAD to come from nothing. They couldn't have evolved gradually by slight modification of existing systems, but the example above is just one example that completely discredits this notion.
The second talking point of his is the idea of genetic entropy. SfT thinks that the genome is somehow under influence of the second law of thermodynamics so it should break down over time, even though the genome is part of an open system and it doesn’t have anything to do with the “order” of a system. Thermodynamic entropy has to do with the amount of energy that is able to do work. This doesn't apply to the "order" or "disorder" of DNA.
The man behind genetic entropy is John Sanford, who is the scientist that co-invented the gene gun, which makes him a respectable scientists in the field of plant genetics. Though, that doesn’t mean all of his ideas are respectable. There are many scientists with ground breaking ideas like Lynn Margulis (Carl Sagan’s first wive) who proposed endosymbiosis, but she was also an HIV denials and 9/11 truther. Likewise Sanford has also some wacky ideas that aren’t supported, like genetic entropy. Sanford claims that genomes are deteriorating over time. This is because in every individual of every generation, there are numerous small deleterious mutations, a good fraction of which have almost no influence on fitness such that selection is ineffective to weed them out. Thus these small harmful mutations will accumulate due to genetic drift, generation by generation. This would inevitably deteriorate the genome over time and populations could never increase in fitness. They will always decrease in fitness. You might think “what about beneficial mutations? Wouldn't they cause an increase in fitness and counter act this "genetic entropy"?” Well, John Sanford simply dismisses the influence of of beneficial mutations, because he thinks that they are too rare and practically all of them are also effectively invisible to selection. He also claims that because of this, beneficial mutations cannot play the role in evolution according to the scientific consensus. More on that later.
One of the things that discredits genetic entropy is by pointing out that most mutations are completely neutral since most stretches of the genome is non-functional. But then, of course, SfT brings up ENCODE to claim that 80% of the human is functional and not junk DNA. However, most of the human genome is actually not functional (don’t have any effect on phenotype or fitness). SciShow did a recent video on this:
ENCODE defined “functional” as any region of DNA that interacted with a binding protein of some sort. It doesn’t matter if anything else happened. If something sticked to the DNA, the DNA is functional according to that ENCODE paper. It’s like (as Jackson Wheat put it) saying that a piece of gum on the ground is functional to your shoe because it can stick to the bottom of your shoe. The genome is a large molecular chain(s) of four different monomers. Simply by change, there would be large sections of DNA that are very similar to protein binding sites. Not entirely identical but similar enough such that proteins will occasionally bind to them and nothing else happens. And when we actually define functional as “DNA that has an effect on fitness or at least the phenotype of the organism” which is more reasonable and useful, we see that the majority of our genome is not functional. But whenever someone explains this SfT he begins to assert that we just assume that the DNA doesn’t have any function simply because we don’t know what it does. That’s not true, we can look at the DNA tell what they do and determine whether they do anything, and we have for most of the genome.
In the functional category we have:
Functional parts of protein coding genes, non-coding RNAs, regulatory sequences, SARs, Origins of replication, centromeres, telomeres, and conserved sequences of unknown function is about 8.7% (essential/functional). Non-functional includes transposable elements, viral DNA, pseudogenes and introns is about 65% (non-functional Junk). The remaining 26.3% is non-conserved intergenic regions of unknown function (probably mostly junk too).
Note: There are very few exceptions such as co-opted viral or pseudo genes that have acquired a new function secondarily, but these are just a fraction of a percentage of all these categories. Creationists will often jump on these exceptions which only account for less than 0.0001% of the genome and use that to claim that the entire genome is functional, which is like cherry picking one cherry on a tree and then claiming that the entire tree is edible. And there are also fractions of a percentage on the functional categories that are actually non-functional, so these small differences won’t change these overall percentages.
So 65% is KNOWN to be non-functional, but we can actually determine how much is likely to be non-functional by other means. There is this thing called genetic load, which sounds similar to the idea of genetic entropy but it is different. It’s real for one thing. The principle behind this is the fact that a population can only tolerate a finite number of deleterious mutations. And it is not like if we went beyond this limit, we would slowly deteriorate (even while increasing our population size) like according to genetic entropy. No, the population couldn’t increase in or maintain its size. We would go into extinction much quicker if we only had four couples (each man related to the same father), like with Noah’s family, because there there is almost nothing that increases the genetic load MORE than incestious inbreeding. But somehow, genetic entropy doesn’t apply with Noah’s family allowing them to produce thousands of offspring in a few generations without deteriorating because….reasons. Anyway..... a paper by Dan Graur published in genome biology and evolution shows how he calculated that we can only have 25% of our genome to be functional at the upper limit. But even this is absurdly generous. A more reasonable value is 10-15%, which is roughly correspond to the previous figure of 8.7% that is known to be functional. Remember, if it was functional over this limit, we would’ve dwindled into extinction a long time ago. Simply of this reason, most of our mutations would be within non-functional regions of the genome and be entirely neutral.
So what more does John Sanford use to support genetic entropy. He often cites various scientists like Michael Lynch and James F. Crow who supposedly all agree that the human genome (or any genome in general) is deteriorating (just like Sanford does):
Sanford also likes to cite his own adapted figure for the frequency distribution of mutations and their effect on fitness from the 1979 paper of Motoo Kimura, here below, to argue for why beneficial mutations are too rare and too low impact to be selected for and counter genetic entropy
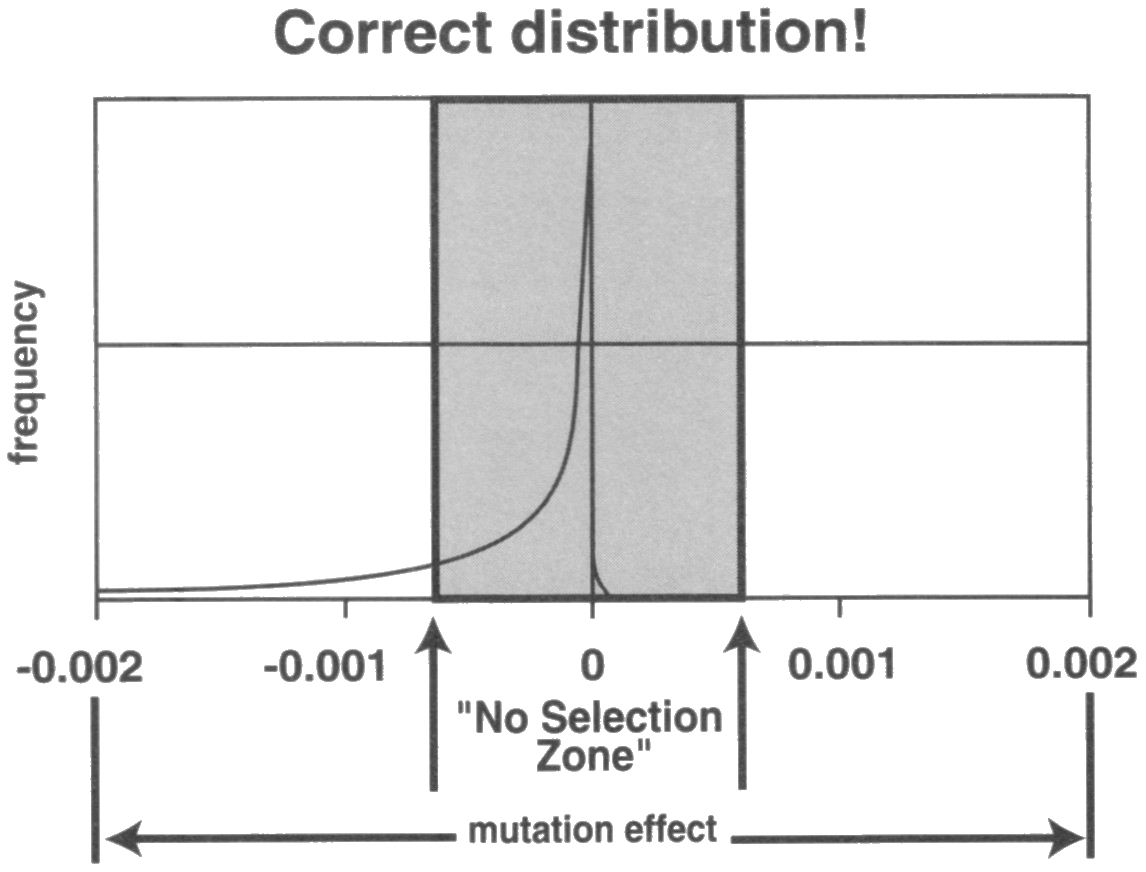
Here is the figure of the original article, figure 1.

His “adapted” figure is fudged in a subtle way. The “no selection zone” is significantly enlarged in his graph, as sanford boundary is closer to the -0.001 mark while the original is closer to the 0.00 mark. There is also the fact that the original doesn’t show anything on the positive (beneficial mutation) side of the fitness scale, while Sanford has added a tiny curve on the right to represent the beneficial mutations, which are apparently infrequent and always inside the “no selection zone”, just as he claims. If John presented this figure as his own speculative hypothesis, to illustrate his ideas, it would be fine. But no, he claimed that this tiny curve also represent what Kimura would have put in for beneficial mutations:
Well….
So what does Sanford have left to speak about? Are there laboratory experiments with populations that show clear genetic entropy (regardless of selection)?
To make matters even worse for Sanford, there are experimental data that actually disproves the idea of genetic entropy. The most famous one is the long Long Term Evolution Experiment by Lenski et al. wherein in one paper they show that even after 50.000 generations, all 12 separate populations still increased in fitness relative to their ancestors following a power law model. This shows that surprisingly fitness can increase indefinitely (or at least for a very long time) even in a constant environment. This flies completely in the face of genetic entropy which says that populations must always decline in fitness over time. In other studies, they also show that beneficial mutations with the same effect occur independently in different populations. (see citations 15 and 16) So these beneficial mutations aren’t too rare nor are they invisible to selection. And of course, the most famous beneficial mutation of the LTEE experiment led to a completely new phenotype in one of the 12 populations. The ability to utilise citrate when oxygen is present.
Now creationist like SfT like to downplay the significance behind this mutations by claiming that it already could digest citrate so it is not “new information” and sometimes go as far to claim that it is actually a decrease in information (SfT specifically said it is a loss of "control") since it now lost its specificity to express these genes only when no oxygen is present. Now it is "unspecific" or "uncontrolled" such that the genes are active all the time. But that is all wrong. The ability to digest citrate under aerobic conditions wasn’t present before, so the phenotype is NEW by any reasonable definition of what count as new. It is also not the case that genetic information was lost since the mutation that was responsible for this new phenotype was a duplication. Well, now you can already hear the creationists yelling “duplications aren’t new information!!!”, but this duplication DID create something new, see diagram here below
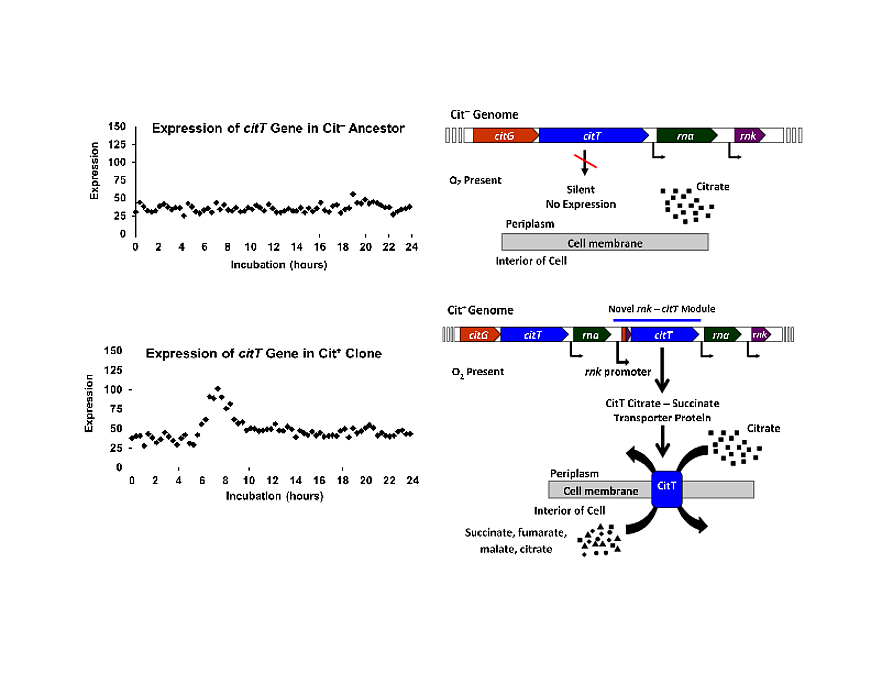
Picture from this
The section that was duplicated included the CitT (the gene which produces a membrane transporter that allows citrate into the cell) at one end and the RNK promotor (which expresses the RNK gene when oxygen is present). The duplication was tandem, meaning the duplicated section was directly put next to the original section. This ended up with a duplicated CitT gene being controlled under a duplicated RNK promotor. Notice that the original genes are still there and still being controlled by the same promoters. There is still a CitT gene controlled by a promoter that is activated when no oxygen is present and there is still an RNK gene controlled by a promoter that activates when oxygen is present. Hence NOTHING WAS LOST. What was gained was a novel regulatory module.
First you had these regulatory modules:
1. Promotor (activate when no oxygen is present) —> CitT
2. Promotor (activate when oxygen is present) —> RNK
After the duplication, you have:
1. Promotor (activate when no oxygen is present) —> CitT
2. Promotor (activate when oxygen is present) —> RNK
3. promotor (activate when oxygen is present) —> CitT
It’s an novel (new) module. No specificity was lost, no loss of control. Control and specificity was gained at the molecular level and a novelty was gained at the phenotype level.
Now I am done. I think that this pretty much shows that Genetic Entropy is complete bollocks as well as other talking points of creationists like “there is no junk DNA” and “no new information”. The sum of the sources are here below.
SOURCES:
1. Stevens, Raymond C., et al. "The GPCR Network: a large-scale collaboration to determine human GPCR structure and function." Nature reviews Drug discovery 12.1 (2013): 25.
2. Sandwalk: What's In Your Genome? - The Pie Chart
3. Sandwalk: Theme: Genomes & Junk DNA
4. Graur, Dan. "An upper limit on the functional fraction of the human genome." Genome biology and evolution 9.7 (2017): 1880-1885.
5. http://sandwalk.blogspot.com/2017/07/revisiting-genetic-load-argument-with.html
6. http://sandwalk.blogspot.com/2009/11/genetic-load-neutral-theory-and-junk.html
7. Rands, Chris M., et al. "8.2% of the human genome is constrained: variation in rates of turnover across functional element classes in the human lineage." PLoS genetics 10.7 (2014): e1004525.
8. Dr. Scott Buchanan "Letters to creationists" Junk_DNA_Design
9. Crow, James F. "The high spontaneous mutation rate: is it a health risk?." Proceedings of the National Academy of Sciences 94.16 (1997): 8380-8386.
10. Lynch, Michael. "Rate, molecular spectrum, and consequences of human mutation." Proceedings of the National Academy of Sciences 107.3 (2010): 961-968.
11. Dr. Scott Buchanan "Letters to creationists" Gen_Entropy
12. Gerard Jellison - Amazon Customer Review for the book "Genetic Entropy" by J. Sanford
13. Simonsen, Lone, et al. "Pandemic versus epidemic influenza mortality: a pattern of changing age distribution." Journal of infectious diseases 178.1 (1998): 53-60.
14. Lenski, Richard E., et al. "Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with Escherichia coli." Proc. R. Soc. B 282.1821 (2015): 20152292.
15. Cooper, Tim F., Daniel E. Rozen, and Richard E. Lenski. "Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli." Proceedings of the National Academy of Sciences 100.3 (2003): 1072-1077.
16. Cooper, Tim F., et al. "Expression profiles reveal parallel evolution of epistatic interactions involving the CRP regulon in Escherichia coli." PLoS Genetics 4.2 (2008): e35.
17. Blount, Zachary D., et al. "Genomic analysis of a key innovation in an experimental Escherichia coli population." Nature 489.7417 (2012): 513.
18. A Duplication Mutation and a New Arrangement: E.coli Cit+ phenotype
JAQing off (short for "Just-asking-questions")
It’s a way to phrase claims as questions in such a way that completely disregards the answers that could be given and is only intended to influence the views of spectators. For example, if an 9/11 truther asks questions about how you can explain their perceived evidence of a controlled demolition, it implies that IF you or someone else is unable to answer the question on the spot then they are seemingly winning the point automatically, hence it WAS evidence of a controlled demolition. It’s a clever way to shift the burden of proof. So SfT often asks questions with claims and assertion hidden within them. That's one thing to watch out for. SfT also employs changing topic constantly by asking new questions immediately after previous one is answers and once he is pinned into a corner (the opponent won't let him change the subject), he apparently begins to freak out and starts spouting a string of nonsense all at once just to flabbergast the opponent and distract them with this steaming pile of BS. A discussion with him would be quite some experience. And I believe that his tactic would work more effectively with people who aren’t prepared to answer his loaded/leading questions, so here I have prepared some answers/rebuttals to his questions/assertions.
One of his main talking points is new genetic information, in which he seems to define as “new functional DNA popping out of nothing”. Note the “out of nothing” caveat. It doesn’t matter if you have a completely new gene forming from non-coding DNA which previously didn’t do anything, it doesn’t count as new information since the template was already there. Of course, Evolution doesn’t entail nor require that things pop out of thing air. Evolution is more like tinkering. Rather than inventing new things it uses old stuff and modifies it to make the new. A good example of this is the GPCR (G-coupled Protein receptors) superfamily. They bind to molecules outside the cell and when that happens they activate an internal signal transduction pathway. It is an extremely fast way for cells to respond to external stimuli. The first GPCR got duplicated, and the duplicated version got duplicated in turn, and so forth, creating this protein super family.
Each duplicated version was slightly modified such that they would react to different molecules. This produced a wide variety of different GPCRs with completely different functions in many systems. There are those that bind to hormones, regulate the immune system and inflammation, some are involved in our sense of smell and taste because they bind to flavour and odour molecules. Some GPCRs even got modified such that they could react to light and are now the opsins in the rod cells of our eyes. It's rather odd that creationists are asking us to show new information, i.e. show how DNA can pop into existence from nothing.
That’s the creationists view, God making things from nothing. So they should be asking this question to themselves, not us.
It also shows that they have this conception that these biological systems just HAD to come from nothing. They couldn't have evolved gradually by slight modification of existing systems, but the example above is just one example that completely discredits this notion.
The second talking point of his is the idea of genetic entropy. SfT thinks that the genome is somehow under influence of the second law of thermodynamics so it should break down over time, even though the genome is part of an open system and it doesn’t have anything to do with the “order” of a system. Thermodynamic entropy has to do with the amount of energy that is able to do work. This doesn't apply to the "order" or "disorder" of DNA.
The man behind genetic entropy is John Sanford, who is the scientist that co-invented the gene gun, which makes him a respectable scientists in the field of plant genetics. Though, that doesn’t mean all of his ideas are respectable. There are many scientists with ground breaking ideas like Lynn Margulis (Carl Sagan’s first wive) who proposed endosymbiosis, but she was also an HIV denials and 9/11 truther. Likewise Sanford has also some wacky ideas that aren’t supported, like genetic entropy. Sanford claims that genomes are deteriorating over time. This is because in every individual of every generation, there are numerous small deleterious mutations, a good fraction of which have almost no influence on fitness such that selection is ineffective to weed them out. Thus these small harmful mutations will accumulate due to genetic drift, generation by generation. This would inevitably deteriorate the genome over time and populations could never increase in fitness. They will always decrease in fitness. You might think “what about beneficial mutations? Wouldn't they cause an increase in fitness and counter act this "genetic entropy"?” Well, John Sanford simply dismisses the influence of of beneficial mutations, because he thinks that they are too rare and practically all of them are also effectively invisible to selection. He also claims that because of this, beneficial mutations cannot play the role in evolution according to the scientific consensus. More on that later.
One of the things that discredits genetic entropy is by pointing out that most mutations are completely neutral since most stretches of the genome is non-functional. But then, of course, SfT brings up ENCODE to claim that 80% of the human is functional and not junk DNA. However, most of the human genome is actually not functional (don’t have any effect on phenotype or fitness). SciShow did a recent video on this:
ENCODE defined “functional” as any region of DNA that interacted with a binding protein of some sort. It doesn’t matter if anything else happened. If something sticked to the DNA, the DNA is functional according to that ENCODE paper. It’s like (as Jackson Wheat put it) saying that a piece of gum on the ground is functional to your shoe because it can stick to the bottom of your shoe. The genome is a large molecular chain(s) of four different monomers. Simply by change, there would be large sections of DNA that are very similar to protein binding sites. Not entirely identical but similar enough such that proteins will occasionally bind to them and nothing else happens. And when we actually define functional as “DNA that has an effect on fitness or at least the phenotype of the organism” which is more reasonable and useful, we see that the majority of our genome is not functional. But whenever someone explains this SfT he begins to assert that we just assume that the DNA doesn’t have any function simply because we don’t know what it does. That’s not true, we can look at the DNA tell what they do and determine whether they do anything, and we have for most of the genome.
In the functional category we have:
Functional parts of protein coding genes, non-coding RNAs, regulatory sequences, SARs, Origins of replication, centromeres, telomeres, and conserved sequences of unknown function is about 8.7% (essential/functional). Non-functional includes transposable elements, viral DNA, pseudogenes and introns is about 65% (non-functional Junk). The remaining 26.3% is non-conserved intergenic regions of unknown function (probably mostly junk too).
Note: There are very few exceptions such as co-opted viral or pseudo genes that have acquired a new function secondarily, but these are just a fraction of a percentage of all these categories. Creationists will often jump on these exceptions which only account for less than 0.0001% of the genome and use that to claim that the entire genome is functional, which is like cherry picking one cherry on a tree and then claiming that the entire tree is edible. And there are also fractions of a percentage on the functional categories that are actually non-functional, so these small differences won’t change these overall percentages.
So 65% is KNOWN to be non-functional, but we can actually determine how much is likely to be non-functional by other means. There is this thing called genetic load, which sounds similar to the idea of genetic entropy but it is different. It’s real for one thing. The principle behind this is the fact that a population can only tolerate a finite number of deleterious mutations. And it is not like if we went beyond this limit, we would slowly deteriorate (even while increasing our population size) like according to genetic entropy. No, the population couldn’t increase in or maintain its size. We would go into extinction much quicker if we only had four couples (each man related to the same father), like with Noah’s family, because there there is almost nothing that increases the genetic load MORE than incestious inbreeding. But somehow, genetic entropy doesn’t apply with Noah’s family allowing them to produce thousands of offspring in a few generations without deteriorating because….reasons. Anyway..... a paper by Dan Graur published in genome biology and evolution shows how he calculated that we can only have 25% of our genome to be functional at the upper limit. But even this is absurdly generous. A more reasonable value is 10-15%, which is roughly correspond to the previous figure of 8.7% that is known to be functional. Remember, if it was functional over this limit, we would’ve dwindled into extinction a long time ago. Simply of this reason, most of our mutations would be within non-functional regions of the genome and be entirely neutral.
So what more does John Sanford use to support genetic entropy. He often cites various scientists like Michael Lynch and James F. Crow who supposedly all agree that the human genome (or any genome in general) is deteriorating (just like Sanford does):
However, Sanford is misrepresenting the views of these scientists. Crow, Lynch and other scientists who are similarly quoted by Sanford are concerned with what is called “Relaxed natural selection”. In order for natural selection to fully operate, populations must produce lots of offspring only some of which would survive and reproduce. But when there are only few offspring per individual and each offspring is kept alive through artificial means, there is little opportunity for deleterious mutations to be purged or beneficial mutation to be rewarded and the fitness of the population declines for this reason. When we observe populations with large individuals and differential reproduction, fitness holds up just fine due to selection having full effect. But when selection is shut down, fitness and genomes deteriorate. And this is what Lynch and Crow are publishing about regarding human populations in industrialised societies which have few offspring per family and each offspring largely being kept alive by medial intervention.J. Sanford said:“Subsequently, they realise that genetic information is currently being lost, which must eventually result in reduced fitness of our species. This decline unfitness is believed to be occurring at 1-2% per generation (Crow 1997)”
“The most definitive findings were published in 2010 in the Proceedings of the National Academy of Science by Lynch. That paper indicates human fitness is declining at 3–5% per generation.”
[url=http://www.pnas.org/content/94/16/8380 said:James F. Crow[/url]"]“However efficient natural selection was in eliminating harmful mutations in the past, it is no longer so in much of the world. In the wealthy nations, natural selection for differential mortality is greatly reduced. A newborn infant now has a large probability of surviving past the reproducing years. There are fertility differences, to be sure, but they are clearly not distributed in such a way as to eliminate mutations efficiently. Except for pre-natal mortality, natural selection for effective mutation removal has been greatly reduced.”
But this doesn’t support Sanford’s claims. Sanford is specifically claiming that even natural selection cannot prevent genetic entropy. Obviously, the work of Lynch and Crow specifically apply to situations without natural selection, but the way Sanford cites these scientists is grossly misleading, such that it sounds like they are saying that the human genome is deteriorating regardless of selection. And there are other researches that shows evidence against the notion that humans are deteriorating due to a lack of selection, which is obviously ignored by Sanford and his ilk.[url=https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2824313/ said:Michael Lynch[/url]"]“the mean phenotypes of the residents of industrialised nations are likely to be rather different in just two or three centuries, with significant incapacitation at the morphological, physiological, and neurobiological levels. Ironically, the genetic future of mankind may reside predominantly in the gene pools of the least industrialised segments of society.”
Sanford also likes to cite his own adapted figure for the frequency distribution of mutations and their effect on fitness from the 1979 paper of Motoo Kimura, here below, to argue for why beneficial mutations are too rare and too low impact to be selected for and counter genetic entropy
Here is the figure of the original article, figure 1.
His “adapted” figure is fudged in a subtle way. The “no selection zone” is significantly enlarged in his graph, as sanford boundary is closer to the -0.001 mark while the original is closer to the 0.00 mark. There is also the fact that the original doesn’t show anything on the positive (beneficial mutation) side of the fitness scale, while Sanford has added a tiny curve on the right to represent the beneficial mutations, which are apparently infrequent and always inside the “no selection zone”, just as he claims. If John presented this figure as his own speculative hypothesis, to illustrate his ideas, it would be fine. But no, he claimed that this tiny curve also represent what Kimura would have put in for beneficial mutations:
But what does Kimura actually say in the article?Sanford said:“In Kimura’s figure, he does not show any mutations to the right of zero – i.e. there are zero beneficial mutations shown. He obviously considered beneficial mutations so rare as to be outside of consideration.”
“So selection could never favor any such beneficial mutations, and they would essentially all drift out of the population. No wonder that Kimura preferred not to represent the distribution of the favorable mutations!”
"Kimura does not show the beneficial distribution, which is essential to the question of net gain versus net loss! When I show the beneficial distribution (while Kimura did not do this, I suspect he would have drawn it much as I did), anyone can see the problem: the vast majority of beneficial mutations will be un-selectable"
Well….
Thus Kimura actually believed that there would be selectable beneficial mutations that compensate for the accumulation of deleterious mutations. So if he did draw a line for beneficial mutations, the curve would extend beyond the non selection zone. The reason why Kimura omitted beneficial mutations in his figure is that, in his primitive mathematical model, the presence of any beneficial mutation would make it blow up, i.e. he omitted them NOT because (as Sanford claims) Kimura believe they were negligible, but because their effect was too strong. (See citations: 11 and 12)[url=https://pdfs.semanticscholar.org/4dd2/88a00d352fd6e7781763a4e26f373f30fc3e.pdf said:Motoo Kimura[/url]"]“Note that in this formulation, we disregard beneficial mutants, and restrict our consideration only to deleterious and neutral mutations. Admittedly, this is an oversimplification, but as I shal show later, a model assuming that beneficial mutations also arise at a constant rate independent of environmental changes leads to unrealistic results.”
“Under the present model, effectively neutral, but, in fact, very slightly deleterious mutants accumulate continuously in every species. The selective disadvantage of such mutant (in terms of an individual’s survival and reproduction - i.e., in Darwinian fitness) is likely to be of the order of 10^-5 or less, but with 10^4 loci per genome coding for various proteins and each accumulating the mutants at the rate of 10^-6 per generation, the rate of loss of fitness per generation may amount to 10^-7 per generation. Whether such a small rate of deterioration in fitness constitutes a threat to the survival and welfare of the species (not to the individual) is a moot point, but this can easily be taken care of by adaptive gene substitutions that must occur from time to time, say once every few hundred generations.”
So what does Sanford have left to speak about? Are there laboratory experiments with populations that show clear genetic entropy (regardless of selection)?
The answer seems to be “no” at least not most experiments. But we do have an interesting caveat here. Genomes are relentlessly deteriorating, yet at the same time it is happening so slowly that we cannot detect it? Notice that the “slowness” of entropy is added just to explain away how species could have survived thousands of years of genetic entropy (especially considering that they all started from just an incestious gene pool right after the flood 4000 years ago). Trying to have your cake and eat it too it seems. About the “downward decay curves” that he cites in his book, I have no better response to this than Dr. Scott Buchanan.[url=https://creation.com/genetic-entropy said:John Sanford[/url]"]
“It is true that most lab experiments do not show clear degeneration. But Scott should realize that anything alive today must have been degenerating slowly enough to still be here, even in a young earth scenario. All three of the downward decay curves I show in my book indicate that degeneration slows dramatically as it becomes more advanced. If a species is alive today and has been around for thousands of years, the rate of degeneration must be very slow (too subtle to measure in most cases). Obviously, genetic degeneration is not going to be clearly visible in most lab experiments.”
In his book (fourth edition), Sanford makes an attempt to support his claims with actual data (emphasis on "attempt"). He compared his figure 14 (results of Sanford’s mendel’s accountant algorithm, that models his genetic entropy, showing a fitness decline over time) and compares it to figure 15, the data of is comes from influenza viruses from the paper by Simonsen et al a rare example of Sanford referencing to real world data. He presents that figure as - “Actual biological data, showing mutation accumulation and fitness decline in human influenza virus”.[url=https://letterstocreationists.wordpress.com/gen_entropy/ said:Scott Buchanan[/url]"]"The claim here is that genomes are relentlessly deteriorating, but we cannot detect it. I’m sure that genetics simulation programs can be adjusted to show that the fitness of all sorts of organisms, from bacteria to elephants, starting from some idealized mutation-free state, could fall dramatically from 4000 B.C to say around 500 B.C., then almost level out. I do not find that compelling support for genetic entropy."
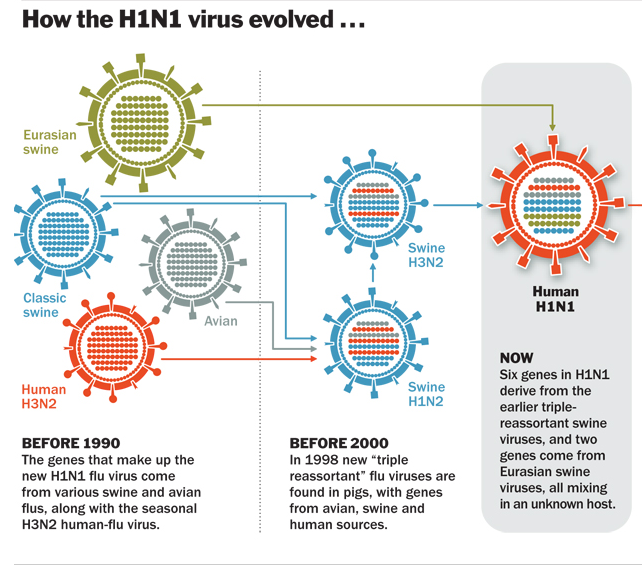
But this is, just like with the Kimura figure, completely misrepresenting the original figure 1 from Simonsen et al. That figure doesn’t quantify pathogenicity nor fitness like Sanford is claiming. As Gerard Jellison said in his review on Amazon:John Sanford said:“This graph illustrates the pathogenicity (i.e., fitness) of the H1N1 strain, as well as the other two pandemic-causing strains during the last century.”
[url=https://www.amazon.com/gp/customer-reviews/R3SC959M9IONT8/ref=cm_cr_arp_d_rvw_ttl?ie=UTF8&ASIN=0981631614 said:Gerard Jellison[/url]"]“…the figure shows a ratio: the number of influenza-caused deaths of people under 65, divided by the total number of deaths. The point of the paper is that the flu epidemic of 1918-1919 was unusual in that it killed mostly young people. The authors studied how this ratio varied with time, and modeled the results in terms of different immunity in the two age cohorts. Thus, the curves in Sanford’s plot merely show that, as time goes on, flu strains kill a greater proportion of elderly people. This makes sense, since young people have more vigorous immune systems [….] Sanford’s Figure 15 is invalid, since it does not illustrate declining viral “fitness” at all. This blunder is an absurd misuse and/or misunderstanding of scientific data.”
To make matters even worse for Sanford, there are experimental data that actually disproves the idea of genetic entropy. The most famous one is the long Long Term Evolution Experiment by Lenski et al. wherein in one paper they show that even after 50.000 generations, all 12 separate populations still increased in fitness relative to their ancestors following a power law model. This shows that surprisingly fitness can increase indefinitely (or at least for a very long time) even in a constant environment. This flies completely in the face of genetic entropy which says that populations must always decline in fitness over time. In other studies, they also show that beneficial mutations with the same effect occur independently in different populations. (see citations 15 and 16) So these beneficial mutations aren’t too rare nor are they invisible to selection. And of course, the most famous beneficial mutation of the LTEE experiment led to a completely new phenotype in one of the 12 populations. The ability to utilise citrate when oxygen is present.
Now creationist like SfT like to downplay the significance behind this mutations by claiming that it already could digest citrate so it is not “new information” and sometimes go as far to claim that it is actually a decrease in information (SfT specifically said it is a loss of "control") since it now lost its specificity to express these genes only when no oxygen is present. Now it is "unspecific" or "uncontrolled" such that the genes are active all the time. But that is all wrong. The ability to digest citrate under aerobic conditions wasn’t present before, so the phenotype is NEW by any reasonable definition of what count as new. It is also not the case that genetic information was lost since the mutation that was responsible for this new phenotype was a duplication. Well, now you can already hear the creationists yelling “duplications aren’t new information!!!”, but this duplication DID create something new, see diagram here below
Picture from this
The section that was duplicated included the CitT (the gene which produces a membrane transporter that allows citrate into the cell) at one end and the RNK promotor (which expresses the RNK gene when oxygen is present). The duplication was tandem, meaning the duplicated section was directly put next to the original section. This ended up with a duplicated CitT gene being controlled under a duplicated RNK promotor. Notice that the original genes are still there and still being controlled by the same promoters. There is still a CitT gene controlled by a promoter that is activated when no oxygen is present and there is still an RNK gene controlled by a promoter that activates when oxygen is present. Hence NOTHING WAS LOST. What was gained was a novel regulatory module.
First you had these regulatory modules:
1. Promotor (activate when no oxygen is present) —> CitT
2. Promotor (activate when oxygen is present) —> RNK
After the duplication, you have:
1. Promotor (activate when no oxygen is present) —> CitT
2. Promotor (activate when oxygen is present) —> RNK
3. promotor (activate when oxygen is present) —> CitT
It’s an novel (new) module. No specificity was lost, no loss of control. Control and specificity was gained at the molecular level and a novelty was gained at the phenotype level.
Now I am done. I think that this pretty much shows that Genetic Entropy is complete bollocks as well as other talking points of creationists like “there is no junk DNA” and “no new information”. The sum of the sources are here below.
SOURCES:
1. Stevens, Raymond C., et al. "The GPCR Network: a large-scale collaboration to determine human GPCR structure and function." Nature reviews Drug discovery 12.1 (2013): 25.
2. Sandwalk: What's In Your Genome? - The Pie Chart
3. Sandwalk: Theme: Genomes & Junk DNA
4. Graur, Dan. "An upper limit on the functional fraction of the human genome." Genome biology and evolution 9.7 (2017): 1880-1885.
5. http://sandwalk.blogspot.com/2017/07/revisiting-genetic-load-argument-with.html
6. http://sandwalk.blogspot.com/2009/11/genetic-load-neutral-theory-and-junk.html
7. Rands, Chris M., et al. "8.2% of the human genome is constrained: variation in rates of turnover across functional element classes in the human lineage." PLoS genetics 10.7 (2014): e1004525.
8. Dr. Scott Buchanan "Letters to creationists" Junk_DNA_Design
9. Crow, James F. "The high spontaneous mutation rate: is it a health risk?." Proceedings of the National Academy of Sciences 94.16 (1997): 8380-8386.
10. Lynch, Michael. "Rate, molecular spectrum, and consequences of human mutation." Proceedings of the National Academy of Sciences 107.3 (2010): 961-968.
11. Dr. Scott Buchanan "Letters to creationists" Gen_Entropy
12. Gerard Jellison - Amazon Customer Review for the book "Genetic Entropy" by J. Sanford
13. Simonsen, Lone, et al. "Pandemic versus epidemic influenza mortality: a pattern of changing age distribution." Journal of infectious diseases 178.1 (1998): 53-60.
14. Lenski, Richard E., et al. "Sustained fitness gains and variability in fitness trajectories in the long-term evolution experiment with Escherichia coli." Proc. R. Soc. B 282.1821 (2015): 20152292.
15. Cooper, Tim F., Daniel E. Rozen, and Richard E. Lenski. "Parallel changes in gene expression after 20,000 generations of evolution in Escherichia coli." Proceedings of the National Academy of Sciences 100.3 (2003): 1072-1077.
16. Cooper, Tim F., et al. "Expression profiles reveal parallel evolution of epistatic interactions involving the CRP regulon in Escherichia coli." PLoS Genetics 4.2 (2008): e35.
17. Blount, Zachary D., et al. "Genomic analysis of a key innovation in an experimental Escherichia coli population." Nature 489.7417 (2012): 513.
18. A Duplication Mutation and a New Arrangement: E.coli Cit+ phenotype


 R His response in a nutshell: For the most part, it is the same stuff re-stated that has already been refuted by me in this thread or in the google hangouts chat. Repeating your claims without while omitting the the counter points doesn't count as a response. Regarding the parts that are new, he mentions some things that are also easily debunked and/or reveals the dishonesty in his position.
R His response in a nutshell: For the most part, it is the same stuff re-stated that has already been refuted by me in this thread or in the google hangouts chat. Repeating your claims without while omitting the the counter points doesn't count as a response. Regarding the parts that are new, he mentions some things that are also easily debunked and/or reveals the dishonesty in his position.