Nesslig20
Active Member
A while back I started a thread on a guy on youtube called "Long Story Short" (I will be using the abbreviation LSS from now on). He makes short animated videos, the art style of which (as I've noted before) look very appealing, but the arguments he makes against evolution are so painfully typical of YEC and ID/creationism. But....AGAIN!!...he isn't a creationist he says. And I take his word for it. The first video that he published on his own channel were about the Cambrian explosion. Jackson Wheat published videos refuting that. Later his videos started appearing on the Discovery Institute. First his video on homology. Jackson refuted those too, which began a series of response videos. And then he started a video on whale evolution. This again started a series of response videos from both sides. Go that thread to see more about that.
Here I want to talk about what he claimed about the papers by:
1. Durrett and Schmidt
2. Behe and Snoke
and
3. Lynch (response paper to Behe and Snoke)
Particularly in this video (the relevant bit starts at 8:41):
He makes several mistakes about that, and Jackson Wheat will publish his response video to this shortly (the script just got completed). However, since we wanted to be as brief as possible, we couldn't include all the relevant details. This is why I am making this new thread here to give the full details for those who are interested. So here we go.
Behe v.s. Lynch
First up, the Behe and Snoke article of 2004. It was published in a legit scientific journal, Protein Science. In that article, they argued that Darwinian mechanisms cannot explain the evolution of protein functions dependent on multiple amino acid residues.
Anyway, the first problem that sticks out from Behe and Snoke’s argument is their stated intention to test whether Darwinian mechanisms are able to produce new multi-residue functions. Since Darwin’s mechanisms only include selection, according to “Darwinian processes” all evolution involves functional intermediate stages. However, Behe and Snoke consider intermediate stages that are neutral. Non-darwinian mechanisms such as drift are indeed important in modern evolutionary biology, but Behe and Snoke’s approach to observe non-darwinian processes in order to test the capabilities of Darwinian mechanisms is just strange to say the least.
Behe and Snoke point out that the function of many proteins are dependent on multiple amino acid residues, which they refer to as “multi-residue” or MR features (or functions). Hence, for a protein to acquire a new function, they have to evolve a new MR feature, which requires multiple mutations. A single mutation just doesn’t cut it - by their reconing. Furthermore, they suppose that this shift in function would come at the cost of losing the original function which may be essential. Hence, the acquisition of a new function can only come about via gene duplication, with one of the copies maintaining the original function such that the other copy is free to mutate. So, Behe and Snoke looked at how a protein would evolve following a gene duplication event, you can see their simplest model in figure 1.
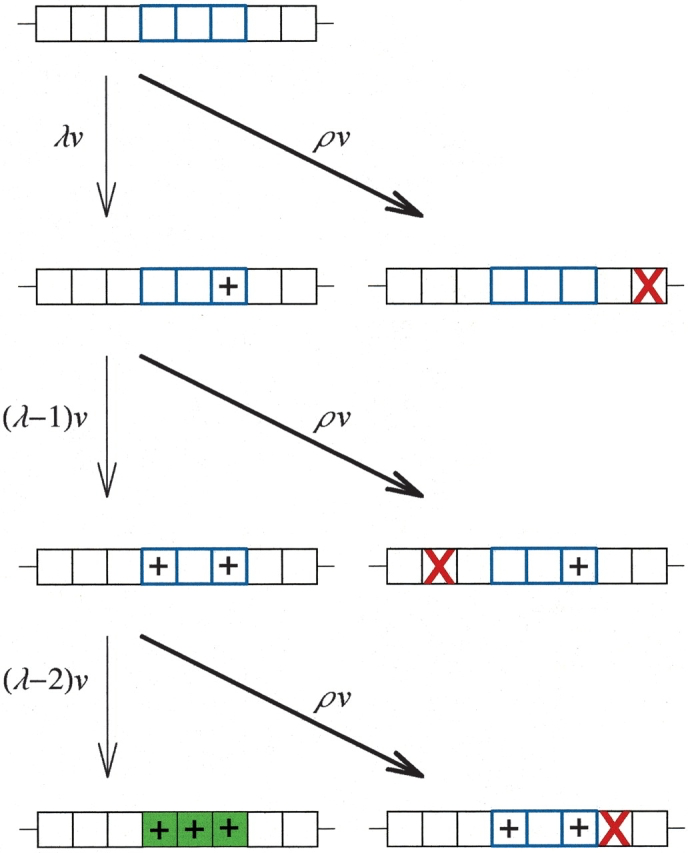
They considered two general outcomes from this setup. In one outcome, one copy undergoes a deleterious mutation (shown as Red X), and it becomes a pseudogene or as they put it a “null allele”. A dead end. The probability of that is given as the product of P (the ratio of all mutations that will result in a “null allele” to all mutations necessary for the novel MR feature) and V (the mutation rate). The alternative outcome is a new gene with a new function. In this model, this would require three specific mutations. The probability of the first correct mutation to occur is the product of λ (the number of loci that needs to be mutated for the formation of the novel MR function) and V (the mutation rate). In this figure there are three mutations needed for the new function to be reached (λ=3), at three loci that are adjacent to each other, but their position doesn’t matter, the probability would be the same if they were positioned differently.
So the whole point is to examine the probability of the second scenario happening, while the first scenario does not happen, within a population with a standard size and experiencing a standard mutation rate. They assume that the population started out with every individual possessing a freshly duplicated gene. And they not only assume that the needed mutations would only COLLECTIVELY (only when all are present) give rise to a new function, but they also assume that each of them INDIVIDUALLY would be detrimental to the original function.
These later assumptions are especially problematic as we will see later. I spare you the formulas given in the paper itself (I also find it difficult to wrap my head around them), but the end value they want to calculate is the number of generations it takes for a new MR function to appear and be fixed in a population (Tfx). Which is depending on, λ, V, P and (two not mentioned so far) N, the population size and S, the selection coefficient. The value of V (mutation rate) they propose is 10^-8 and that S is 0.01. They also assume that P is ~ 1000, since they argue that there are ~ 2800 substitutions that lead to a nonfunctional protein, so (if λ~3) then P is ~2800/~3, which is ~1000.
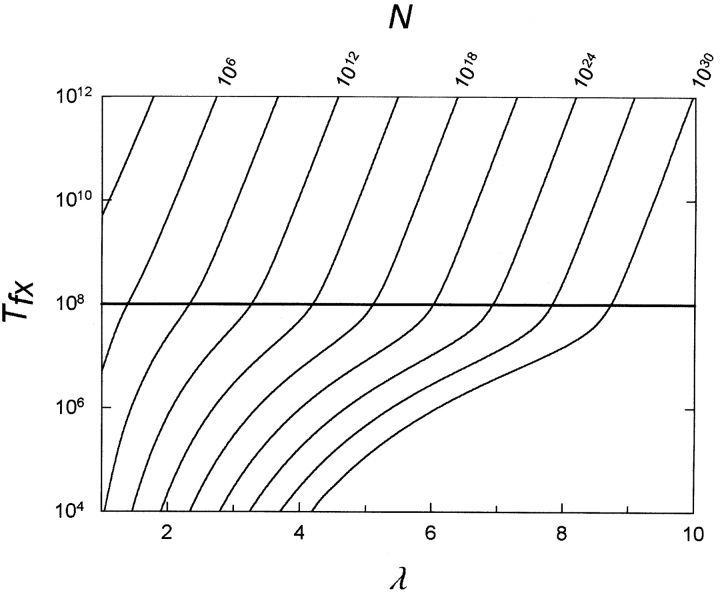
For example, if λ=3, then with a population size of little over 10^11, then the Tfx is about 10^8 generations. To put it simply, if you need 3 mutations to get a new gene with a new function, then even for a population of little more than 100 billion individuals, it still takes on average 100 million generations for the new gene to appear and be fixed. If you want it to happen faster you need to either decrease λ, which is already at 3 in this example. If it is 2, a population size of over 10^9 would still be needed in order for the new functional gene to appear and be fixated within 100 million generations. So the only way you could get a novel MR function in a protein, you need abnormally huge population sizes that exceeds that of most known species. So the conclusion Behe and Snoke draw from this is that the evolution of an novel MR function in a duplicated gene is too improbable within reasonable time frames, and thus “Darwinian processes” are insufficient to explain the origins of new functions that require multiple mutations.
Of course, this conclusion is flawed. Not just because they use the term “darwinian processes” differently compared to how these terms are normally used in evolutionary science (okay, it’s semantics, but still). Behe and Snoke do admit that their estimation could be wrong, but they argue that - if they are wrong - their numbers would actually be underestimates of the real thing. For one, as pointed out before, they have already started with a population with every individual possessing a duplicated gene. So the time it takes for the duplicated gene to spread throughout the population should also be taken into account. That’s fair, however, one of the biggest flaws they made is that they restricted their focus on one of the most difficult pathways one could imagine to produce an adaptive product,
This is where the paper by Michael Lynch comes in, also published in the same journal one year later in 2005. It thoroughly refutes Behe’s and Snoke’s paper.
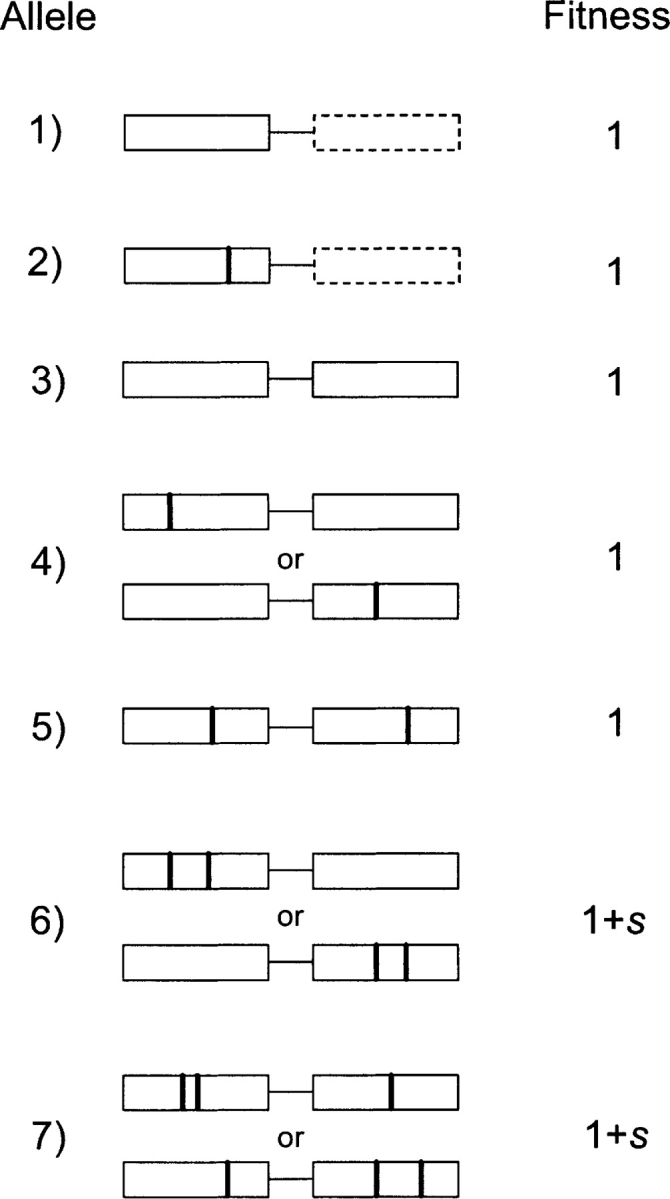
The vertical lines represent the mutations that are needed to change the function. #2 is an example where the mutation happened before the duplication event. You might be thinking that Lynch is wrong to make the assumption of neutrality and that Behe and Snoke are right to assume that each of the mutations towards the new function would be detrimental to the old function. In fact, that’s exactly what Behe and Snoke claimed in their response to Lynch (which LSS also quoted in his video):
As he pointed out, the functionality of proteins are often very tolerant to changes in individual amino acids, which is a form of “robustness”. This fact has been known for a long time, since the 1990s when James Well’s developed the “Alanine Scanning” technique. This involves systematically substituting each amino acid in a protein one-by-one with alanine (the most simplest amino acid), and observing how it affected the protein function. It generally showed that there usually are only a limited number of residues that are REALLY important to the function, while mutations at the other site are mostly tolerable. A more intensive “scanning” process is substituting each site systematically with all of the 20 amino acids (of course not at the same time). Here is an example of HIV-1 protease, shown in the figures below.
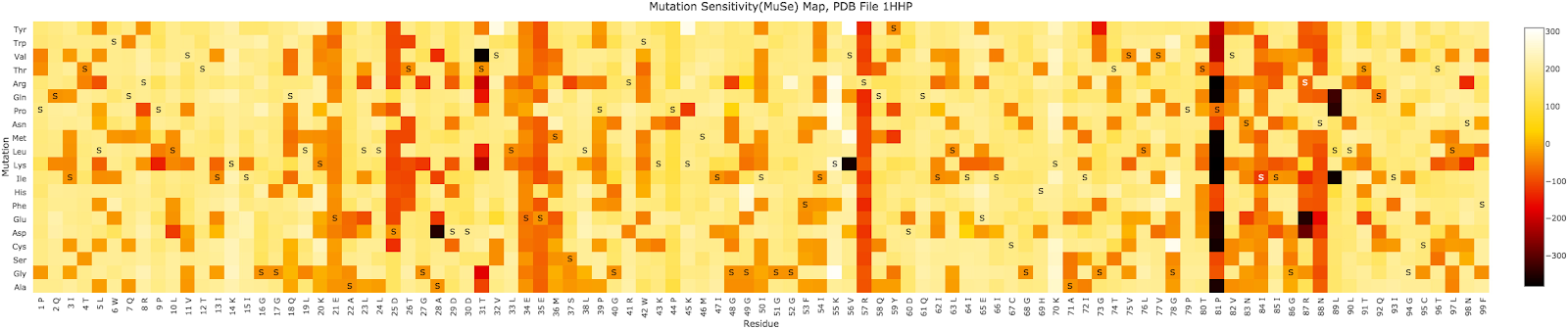
The image (a mutation sensitivity map) shows that most sites are fairly resistant against most aa substitutions, which is why the image on top shows mostly yellow colours, indicating the substitution didn’t have much effect. Only the ones that are rather darkly coloured represent the mutations that would have a strong negative effect on the protein function. So, Behe and Snoke, and thus LSS quoting them, are just wrong when they claim “the majority of amino acid substitutions decrease protein function”.
Lynch also includes another variable called “n”, which is the number of amino acids which could potentially contribute to a new function after a mutation. Behe and Snoke assumed that is only one combination possible that would lead to a new function, so if at minimum 2 mutations are needed then n=2, if there are 3 mutations needed, n=3. Here, Lynch modeled the numbers to be 10 or as high as 50 (which is significant as you will see next). Most of the other parameters were the same as Behe and Snoke (e.g. mutation rate of 10^-8). His results are summarised in this figure.
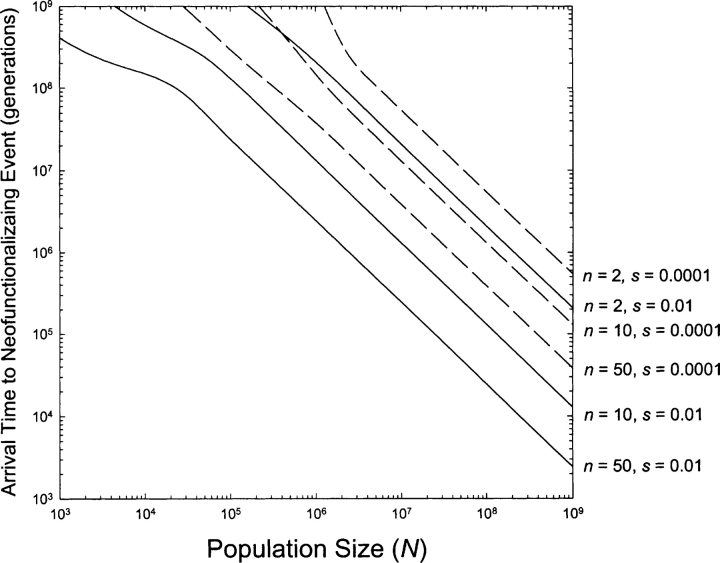
Lynch’s model shows that (when n=50) you would only need a population size of a little more than 1 million in order for a new functional protein to appear and be fixated within 1 million years on average. Contrast this to Behe and Snoke’s model, which would conclude that you need a population size of 1 trillion for the same fixation time of 1 million years, a difference of six orders of magnitude. There are more subtle differences between the models than I mentioned. You can see more problems that have been pointed out by other commentators in the editorial from the journal [or see Lauran Moran’s blog posts 1 and 2 below]. However, I covered the main differences in their assumptions which account for the majority of this discrepancy between the models of Lynch and Behe/Snoke. And, as previously shown, Lynch’s assumption of neutrality is better supported. It should also be stated that Lynch has stated that even his model is still quite restrictive, as there are other possible ways for new genes to evolve a new function following gene duplication that he hasn’t accounted for in his model.
To further support Lynch’s assumptions, there was an laboratory experiment wherein they observed the process of gene duplication and the evolution of a new function that is (though not identical to, bus still) concordant with Lynch’s model. This was documented in the 2012 paper by Nasvall et al. Their observation centered around the gene HisA, which codes for the enzyme that catalyzes the production of histidine. From this gene, they have obtained a mutated version which is, in addition to its original function, is also capable of catalyzing the production of Tryptophan, but very poorly so. While this initial step was artificially induced by the scientists (which is undoubtedly what ID-proponents will point to dismiss this paper outright. The old “See, it had to happen in a lab. It was intelligent design after all!!”). However it shows that mutations which cause a new function to a protein aren’t necessarily detrimental to the original function. Again, supporting Lynch’s assumption of neutrality and that the mutations towards the innovation happened BEFORE duplication, which is starkly different from the model assumed by Behe and Snoke. What they did next is introduce this gene into a species of Salmonella that lacks the normal genes to synthesise histidine and tryptophan, and they grew it in a media without histidine and tryptophan. Hence, the bacteria is entirely reliant on this single gene to obtain both essential amino acids. They also placed this gene such that it was encouraged to be duplicated. Since the tryptophan activity of this gene is rather weak, gene duplication would actually be a beneficial mutation since it increases the production simply by increasing gene dosage (the fact that gene duplication itself can provide a selective advantage is also something that Behe and Snoke didn’t consider). From this point on, they let the bacteria grow for 3000 generations and the results are pretty amazing. What they saw is that in many lineages, the gene got indeed duplicated, but also many lineages experienced mutations that made one duplicated version a lot better at producing tryptophan, while the other was either better at producing only histidine or both. Not only that, similar results happened in multiple lineages independently, see figure below.
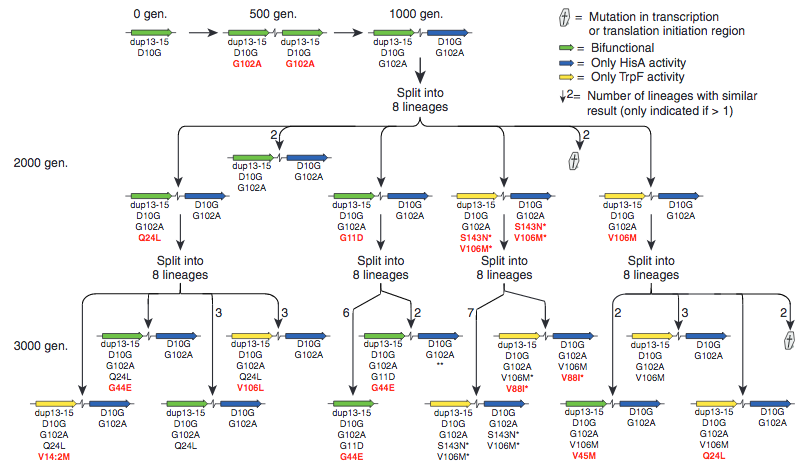
Here blow is a graph that shows the different evolutionary routes these bacteria made regarding how they improved their genes.
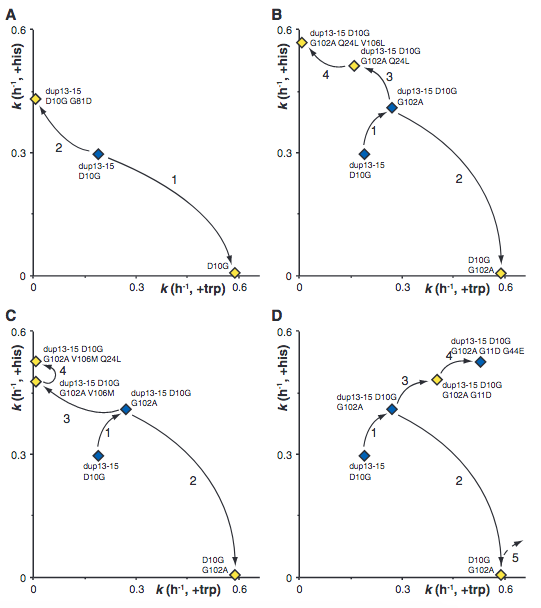
The Y-axis shows how “good” a gene is at producing histidine, and the X-axis shows how “good” it is at producing tryptophan. The 4 graphs show 4 different ways how duplicated genes diverged from one another in both activities (the numbers show how many times these routes were taken independently). Note that there are sometimes different mutations that would lead to roughly the same result. This is concordant with another assumption that Lynch made. That there are multiple combinations of mutations to reach similar results - there are multiple routes to reach similar destinations.
So, in short Lynch is right. Behe and Snoke are wrong.
Behe v.s. Durrett & Schmidt.
Next we have the Durrett & Schmidt paper from 2008. In LSS’ first video on whale evolution, he brought this paper up to argue that the waiting time for “even just two simple beneficial mutations” is too long to have happened within the ~10 million years it took for whales to evolve from being fully terrestrial to fully aquatic. The evolutionists don’t have enough time for even 2 beneficial mutations to have for whale evolution, let alone all the other ones that would also be needed!!
Here is where he went wrong. His first mistakes was saying that the numbers “a few million years” for fruit flies and “200 million years” for humans were about two beneficial mutations, while they were in fact about two NEUTRAL mutations (for fruit flies) and one neutral and one beneficial mutation (for humans). But the bigger problem here is that this paper isn’t about the waiting time for ANY two beneficial mutations. It was about two PRESPECIFIED mutations. In Jackson Wheat’s first rebuttal, he pointed this out, using the analogy of the difference between the odds of ANYONE winning the lottery and a SPECIFIC individual winning the lottery. If there is only one player, then yes, the odds between them are the same, but in a lottery there are a lot of players, so the odds that ONE of them will win is substantially greater than if you calculate the odds of a specific person winning. Which should remind you of the previous topic of Lynch, where it was noted there are many other ways for beneficial mutations to occur. There are even multiple mutations that have the same effect.
In LSS’ response video to Jackson, he made several mistakes regarding this paper. He claimed that the 2008 Durrett and Schmidt paper attempted to disprove the model presented in the 2004 Behe and Snoke paper (which was previously discussed). This just shows that LSS has not read the paper, nor the responses to Durrett and Schmidt written by Behe himself. If he did, he didn’t understand any of it. This wasn’t the goal of Durrett and Schmidt at all. They only mentioned the Behe and Snoke paper more as a side note, while only pointing out a problem they observed additional to those pointed out by Lynch.
In LSS’ response video to Jackson, he wrongly claimed that Durrett and Schmidt narrowed their focus on two specific mutations, because, as he said:
Of course, Behe insists in his second reply that it is very likely deleterious.
LSS strangely highlights this paragraph from Behe, while stating that “the empirical data suggests that the intermediate mutations are not neutral or harmless”. He includes no references to show this empirical data. Neither does Behe here in this paragraph. He assumes (as it says right there) it is likely seriously deleterious to come to his conclusions. We already noted in the previous topic that proteins are often very resistant to aa substitutions, so the assumption of neutrality is actually very plausible. But let’s put a pin on this and return to this later.
The next paragraph LSS highlights is this:
The second to last last thing LSS highlights from Behe’s second reply this:
They pointed out the same thing that I previously did, several times. It’s the same mistake LSS originally made in his video about the Durrett & Schmidt paper. There are multiple possible mutation pairs that could be beneficial, besides one prespecified pair of mutations. This is what Behe refers to as “multiplication of probabilistic recourse”, which - again - was not included in their calculations, since - again - the aim was to calculate the waiting time for one pair of PRESPECIFIED beneficial mutations. Not ANY pair of beneficial mutations. Of course, Behe refused to accept this.
One last thing I want to cover from LSS is this statement:
Now we get to the finale! I have pinned two points for this end paragraph, which are the most important points of contention between Behe and Durrett & Schmidt.
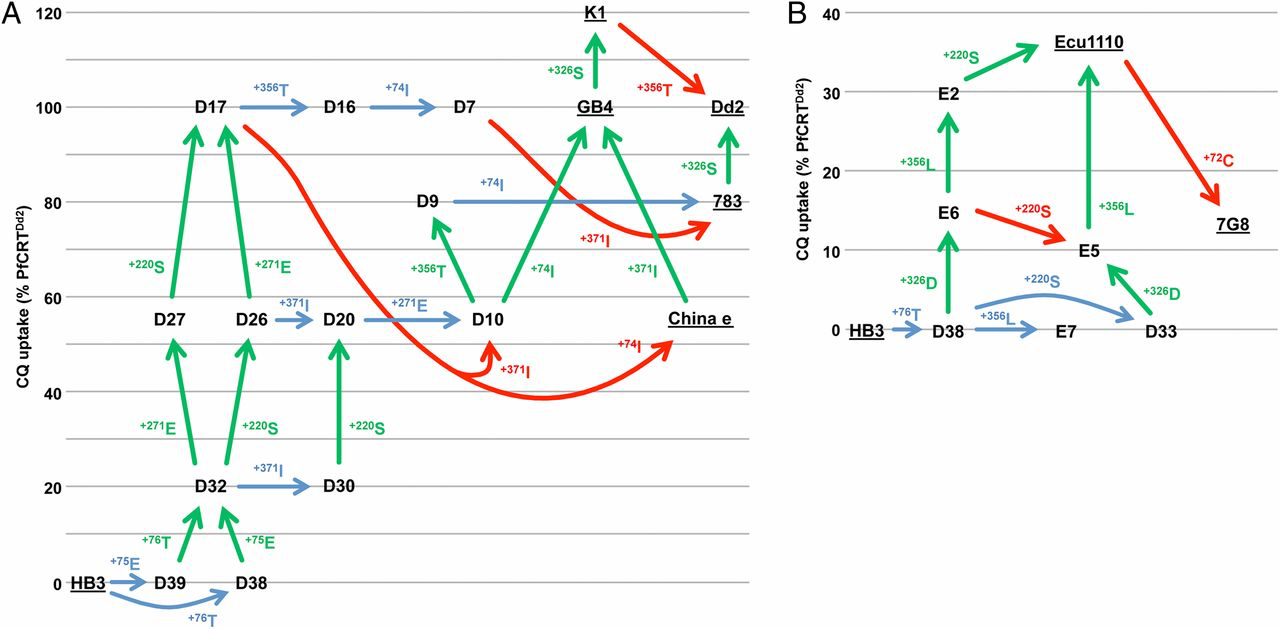
It shows different strains that have different variants of the PfCRT gene, which have different levels of Chloroquine (CQ) uptake capabilities (i.e. how high they are positioned on the figure). The differences between these variants are single aa substitutions, which are shown as codes (i.e +75E). The arrows basically show how chloroquine resistance evolved via a series of strains, each acquiring one specific mutation at a time. The coloured arrows indicate whether the mutation was beneficial (green), detrimental (red) or neutral (blue). Note that many mutations here are neutral, even the ones occurring after the starting point on the bottom left (HB3). Hence, Behe was wrong when he claimed the first mutation is deleterious.
CORRECTION: I have to make a correction after I saw Ration alMind's comments (somewhere else, not here on LoR). I mistakingly took the CQ uptake capacity to be equal to fitness. The coloured lines in the figure denote whether the mutation is neutral/beneficial/detrimental with respect to Chloroquine uptake, but that doesn't mean overall fitness. The paper actually references studies that shows that strains, possessing the mutations for increase CQ uptake, are actually worse-off than the wild-type in the absence of CQ. So there seems to be a trade-off between CQ uptake and the "normal" function of the PfCRT protein (similar to the one described in the BONUS paper).
Furthermore, a paper from 2016 shows how resistance can be achieved by multiple evolutionary routes of 4 mutations, which continuously increased fitness after each mutation. So even a single mutation could confer a slight increase in resistance (although at first very slightly).

The paper also emphasised how important it is for investigating the evolvability of particular phenotypes by modelling different selection pressures, not just looking at the effects of one mutation under a few environments.
There is even a wild variant 106/1, which possesses some of the mutations needed so it isn’t resistant yet, but it is able to become resistant by just one more mutation. Hence, Behe was also wrong when he claimed that the two mutations had to occur at the same time. Not only that, if you look closely at the two possible ways for HB3 to become D32, one path shows the mutations happening with first +75E then 76T, and the other shows first 76T then 76E. So not only don’t they have to be simultaneous, they can also happen in any order, something that even Durrett and Schmidt didn’t account for. You can also see that, since there isn’t just a single series of arrows, there are many ways for evolution to improve CQ uptake. Hence, Behe was also wrong when he only allowed evolution to take one specific way to reach the destination of chloroquine resistance.
It is funny to note that this figure shows the step-by-step evolution of something, which Behe considers too improbable to have evolved. How Behe, the champion of irreducible complexity, can look at this paper and say “yeah, that supports my position” is just beyond me.
This should be also a lesson for many people who accept evolution out there. Natural selection is important, certainly, but it isn’t the only significant aspect of it, not even when it comes to adaptation. As you can see here, some of the necessary intermediate steps are neutral, or sometimes even deleterious. These mutations that are required, but themselves not sufficient, are often dubbed “potentiating” mutations. Selection obviously wouldn’t promote these, but drift makes it possible for some to spread and remain in a population until further mutation occurs that “completes” the phenotype. So not every incremental step HAS to be beneficial for evolution, they can be neutral (or even deleterious). The second lesson is that when we observe, in hindsight, the many mutations that were involved in producing a beneficial trait, that doesn’t mean that these are the ONLY mutations that could have produced a beneficial trait (not even the same beneficial trait) or that they HAD to occur in one particular order. As we have seen, even for the same phenotype (e.g. CQ resistance), there are multiple routes evolution could take and could've taken. I previously described this fallacy with the lottery analogy. This is a common cognitive pitfall that, when people note in hindsight an extremely unlikely event, it makes them think it couldn’t have happened by chance alone. In foresight, most of all extremely unlikely events that you could prescribe will never occur, but since there are so many of these, extremely unlikely events occuring is a mathematical certainty.
EDIT: And a third lesson (also for myself, regarding the correction), whether a mutation is neutral, beneficial or deleterious is often dependent on the environment. Under environmental conditions A, B, C they may be beneficial, but under X, Y and Z, they may be deleterious.
So when someone (creationist or evolutionist) comes to you and says that EVERY incremental step had to infer some advantage, or if they say that X evolved via route Y, route Y was the ONLY way for X to have evolved, please kindly introduce them to this thread for me.
So that’s it. My apologies for the long post. I will leave you all with the citations below, plus a bonus paper (from 2019) that I found while doing research on this. It is on the evolution of drug resistance in Malaria and also on the same PfCRT gene, but here it shows that mutations to this gene can make it resistant to different types of drugs, but not “optimally” for both, i.e. mutations that make it better resistant to one drug makes it less resistance to the other drug as well. A great example of evolutionary trade-offs/constraints. It’s a good read.
CITATIONS
Citations for Behe v.s. Lynch
Main
2004 Behe and Snoke: Simulating evolution by gene duplication of protein features that require multiple amino acid residues
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2286568/
2005 Lynch: Simple evolutionary pathways to complex proteins
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253472/
2005 Behe: A response to Michael Lynch
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253464/
2005 Editorial and position papers
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253483/
Further
1989 High-Resolution Epitope Mapping of hGH-Receptor Interactions by Alanine-Scanning Mutagenesis
https://sci-hub.tw/10.1126/science.2471267
2018 Mutation Sensitivity Maps: Identifying Residue Substitutions That Impact Protein Structure Via a Rigidity Analysis In Silico Mutation Approach
https://www.liebertpub.com/doi/10.1089/cmb.2017.0165?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub 0pubmed
2004 White: Animal drug resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC385418/
Extra: Blog posts by Laurence Moran, which explains the subject more briefly and is easier to understand than the technical literature above.
1. Waiting for multiple mutations: Intelligent Design Creationism v. population genetics
[Explains the Behe and Snoke paper]
https://sandwalk.blogspot.com/2015/12/waiting-for-multiple-mutations.html
2. Waiting for multiple mutations: Michael Lynch v. Michael Behe
[Explains Lynch’s response paper to Behe and Snoke, and also briefly mentions Durrett and Schmidt]
https://sandwalk.blogspot.com/2015/12/waiting-for-multiple-mutations-michael.html
Citations for Behe v.s. Durrett & Schmidt
2008 Durrett and Schmidt: Waiting for Two Mutations: With Applications to Regulatory Sequence Evolution and the Limits of Darwinian Evolution
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2581952/
2009 Behe: Waiting longer for two mutations
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2644969/
2009 Durrett and Schmidt: Reply to Behe
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2644970/
2009 Behe: A roll-up of "Waiting Longer for Two Mutations", Parts 1-5
https://www.discovery.org/a/9611/
2005 A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1173140/
2014 Summers: Diverse mutational pathways converge on saturable chloroquine transport via the malaria parasite’s chloroquine resistance transporter
https://doi.org/10.1073/pnas.1322965111
2016 Combinatorial Genetic Modeling of pfcrt-Mediated Drug Resistance Evolution in Plasmodium falciparum
https://doi.org/10.1093/molbev/msw037
Extra: Blog posts by Laurence Moran, PZ Myers and Kenneth Miller
2012 Moran: Understanding Mutation Rates and Evolution
https://sandwalk.blogspot.com/2012/01/understanding-mutation-rates-and.html
2014 Moran: Taking the Behe challenge!
https://sandwalk.blogspot.com/2014/08/taking-behe-challenge.html
2014 Moran: Flunking the Behe challenge!
https://sandwalk.blogspot.com/2014/08/flunkiung-behe-challenge.html
2014 Moran: CCC's and the edge of evolution
https://sandwalk.blogspot.com/2014/08/cccs-and-edge-of-evolution.html
2014 Moran: Michael Behe's final thoughts on the edge of evolution
https://sandwalk.blogspot.com/2014/08/michael-behes-final-thoughts-on-edge-of.html
2014 Moran: Understanding Michael Behe
https://sandwalk.blogspot.com/2014/08/understanding-michael-behe.html
2014 Moran: On the irrelevance of Michael Behe
https://sandwalk.blogspot.com/2014/12/on-irrelevance-of-michael-behe.html
2016 Moran: Revisiting Michael Behe's challenge and revealing a closed mind
https://sandwalk.blogspot.com/2016/12/revisiting-michael-behes-challenge-and.html
2016 Moran: Targets, arrows, and the lottery fallacy
https://sandwalk.blogspot.com/2016/01/targets-arrows-and-lottery-fallacy.html
2014 PZ Myers: Quote-mined by Casey Luskin!
https://freethoughtblogs.com/pharyngula/2014/07/17/quote-mined-by-casey-luskin/
2014 PZ Myers: Aren’t we all more than a little tired of Michael Behe?
https://freethoughtblogs.com/pharyn...all-more-than-a-little-tired-of-michael-behe/
2007 Kenneth Miller: Falling over the edge
https://www.nature.com/articles/4471055a
2014 Kenneth Miller: Edging towards irrelevance
http://www.millerandlevine.com/evolution/behe-2014/Behe-1.html
BONUS PAPER
2019 Nature News “Malaria parasites fine-tune mutations to resist drugs”
https://www.nature.com/articles/d41586-019-03587-0
2019 Paper: Structure and drug resistance of the Plasmodium falciparum transporter PfCRT
https://doi.org/10.1038/s41586-019-1795-x
Here I want to talk about what he claimed about the papers by:
1. Durrett and Schmidt
2. Behe and Snoke
and
3. Lynch (response paper to Behe and Snoke)
Particularly in this video (the relevant bit starts at 8:41):
He makes several mistakes about that, and Jackson Wheat will publish his response video to this shortly (the script just got completed). However, since we wanted to be as brief as possible, we couldn't include all the relevant details. This is why I am making this new thread here to give the full details for those who are interested. So here we go.
Behe v.s. Lynch
First up, the Behe and Snoke article of 2004. It was published in a legit scientific journal, Protein Science. In that article, they argued that Darwinian mechanisms cannot explain the evolution of protein functions dependent on multiple amino acid residues.
Even though Behe and Snoke are proponents of the idea that an unnamed and mysterious external intelligence is the best explanation for this, they do not provide any alternative mechanism in this paper. There is no mention of “intelligent design”, it’s purely a critique of evolution (or what they refer to as “Darwinian processes”). This is important to note, since this is one paper that the Discovery Institute in their list of "Peer-Reviewed Articles Supporting Intelligent Design" (which I made a video about a while back). However, this particular paper actually doesn’t support ID, EVEN if you assume the contents of the paper is entirely accurate.“Although many scientists assume that Darwinian processes account for the evolution of complex biochemical systems, we are skeptical. Thus, rather than simply assuming the general efficacy of random mutation and selection, we want to examine, to the extent possible, which changes are reasonable to expect from a Darwinian process and which are not. We think the most tractable place to begin is with questions of protein structure. Our approach is to examine pathways that are currently considered to be likely routes of evolutionary development and see what types of changes Darwinian processes may be expected to promote along a particular pathway.”
Anyway, the first problem that sticks out from Behe and Snoke’s argument is their stated intention to test whether Darwinian mechanisms are able to produce new multi-residue functions. Since Darwin’s mechanisms only include selection, according to “Darwinian processes” all evolution involves functional intermediate stages. However, Behe and Snoke consider intermediate stages that are neutral. Non-darwinian mechanisms such as drift are indeed important in modern evolutionary biology, but Behe and Snoke’s approach to observe non-darwinian processes in order to test the capabilities of Darwinian mechanisms is just strange to say the least.
Behe and Snoke point out that the function of many proteins are dependent on multiple amino acid residues, which they refer to as “multi-residue” or MR features (or functions). Hence, for a protein to acquire a new function, they have to evolve a new MR feature, which requires multiple mutations. A single mutation just doesn’t cut it - by their reconing. Furthermore, they suppose that this shift in function would come at the cost of losing the original function which may be essential. Hence, the acquisition of a new function can only come about via gene duplication, with one of the copies maintaining the original function such that the other copy is free to mutate. So, Behe and Snoke looked at how a protein would evolve following a gene duplication event, you can see their simplest model in figure 1.
They considered two general outcomes from this setup. In one outcome, one copy undergoes a deleterious mutation (shown as Red X), and it becomes a pseudogene or as they put it a “null allele”. A dead end. The probability of that is given as the product of P (the ratio of all mutations that will result in a “null allele” to all mutations necessary for the novel MR feature) and V (the mutation rate). The alternative outcome is a new gene with a new function. In this model, this would require three specific mutations. The probability of the first correct mutation to occur is the product of λ (the number of loci that needs to be mutated for the formation of the novel MR function) and V (the mutation rate). In this figure there are three mutations needed for the new function to be reached (λ=3), at three loci that are adjacent to each other, but their position doesn’t matter, the probability would be the same if they were positioned differently.
So the whole point is to examine the probability of the second scenario happening, while the first scenario does not happen, within a population with a standard size and experiencing a standard mutation rate. They assume that the population started out with every individual possessing a freshly duplicated gene. And they not only assume that the needed mutations would only COLLECTIVELY (only when all are present) give rise to a new function, but they also assume that each of them INDIVIDUALLY would be detrimental to the original function.
“The pertinent feature of the model is that multiple changes are required in the gene before the new, selectable feature appears. Changes in these nucleotide positions are assumed to be individually disruptive of the original function of the protein but are assumed either to enhance the original function or to confer a new function once all are in the compatible state. Thus, the mutations would be strongly selected against in an unduplicated gene, because its function would be disrupted and no duplicate would be available to back up the function.”
These later assumptions are especially problematic as we will see later. I spare you the formulas given in the paper itself (I also find it difficult to wrap my head around them), but the end value they want to calculate is the number of generations it takes for a new MR function to appear and be fixed in a population (Tfx). Which is depending on, λ, V, P and (two not mentioned so far) N, the population size and S, the selection coefficient. The value of V (mutation rate) they propose is 10^-8 and that S is 0.01. They also assume that P is ~ 1000, since they argue that there are ~ 2800 substitutions that lead to a nonfunctional protein, so (if λ~3) then P is ~2800/~3, which is ~1000.
Based on these set parameters for V, P and S, they calculated Tfx using different population sizes (N) and different values of λ. The results are shown in figure 6.“An estimate of ρ can be inferred from studies of the tolerance of proteins to amino acid substitution. Although there is variation among different positions in a protein sequence, with surface residues in general being more tolerant of substitution than buried residues, it can be calculated that on average a given position will tolerate about six different amino acid residues and still maintain function (Reidhaar-Olson and Sauer 1988, 1990; Bowie and Sauer 1989; Lim and Sauer 1989; Bowie et al. 1990; Rennell et al. 1991; Axe et al. 1996; Huang et al. 1996; Sauer et al. 1996; Suckow et al. 1996). Conversely, mutations to an average of 14 residues per site will produce a null allele, that is, one coding for a nonfunctional protein. Thus, in the coding sequence for an average-sized protein domain of 200 amino acid residues, there are, on average, 2800 possible substitutions that lead to a nonfunctional protein as a result of direct effects on protein structure or function. If several mutations are required to produce a new MR feature in a protein, then ρ is roughly of the order of 1000. This value for ρ is on the low end used by Walsh (1995), who considered values for ρ up to 10^5. (Walsh, however, defined ρ as the ratio of advantageous-to-null mutations—the inverse of our definition.)”
For example, if λ=3, then with a population size of little over 10^11, then the Tfx is about 10^8 generations. To put it simply, if you need 3 mutations to get a new gene with a new function, then even for a population of little more than 100 billion individuals, it still takes on average 100 million generations for the new gene to appear and be fixed. If you want it to happen faster you need to either decrease λ, which is already at 3 in this example. If it is 2, a population size of over 10^9 would still be needed in order for the new functional gene to appear and be fixated within 100 million generations. So the only way you could get a novel MR function in a protein, you need abnormally huge population sizes that exceeds that of most known species. So the conclusion Behe and Snoke draw from this is that the evolution of an novel MR function in a duplicated gene is too improbable within reasonable time frames, and thus “Darwinian processes” are insufficient to explain the origins of new functions that require multiple mutations.
Of course, this conclusion is flawed. Not just because they use the term “darwinian processes” differently compared to how these terms are normally used in evolutionary science (okay, it’s semantics, but still). Behe and Snoke do admit that their estimation could be wrong, but they argue that - if they are wrong - their numbers would actually be underestimates of the real thing. For one, as pointed out before, they have already started with a population with every individual possessing a duplicated gene. So the time it takes for the duplicated gene to spread throughout the population should also be taken into account. That’s fair, however, one of the biggest flaws they made is that they restricted their focus on one of the most difficult pathways one could imagine to produce an adaptive product,
contrary to their statement:"To support their contention of the implausibility of adaptive protein evolution by Darwinian processes, Behe and Snoke started with an ad hoc non-Darwinian model with a highly restrictive and biologically unrealistic set of assumptions. Such extreme starting conditions guaranteed that the probability of neofunctionalization would be reduced to a minimal level. An alternative approach, adopted here, is to rely on a set of biologically justified premises and an explicit population-genetic framework."
So, it is no surprise that their conclusion was unfavorable to the “Darwinian mechanisms”. They assume that there is only ONE pathway to a new function. You could assume that you need at least 3 mutations to produce a new function, but that doesn’t mean that there is only one combination of 3 mutations that will give rise to a new function. Yeah, mutating sites A, B and C will give a new function, but what about mutating sites D, E and F? Or what about B, C and D? [To preemptively respond to LSS. NO, I am not saying that ALL possible three combinatory mutations could lead to a new function, I am saying that the possible three mutation combination that could lead to a new function isn’t just one.] And what if multiple possible combinations would give rise to the same function or a very similar one? To use an analogy, they assume there is only one road you have to take to a particular destination and that there is only one destination that you can aim for."Our approach is to examine pathways that are currently considered to be likely routes of evolutionary development and see what types of changes Darwinian processes may be expected to promote along a particular pathway."
This is where the paper by Michael Lynch comes in, also published in the same journal one year later in 2005. It thoroughly refutes Behe’s and Snoke’s paper.
Lynch models the possible routes evolution could take to produce a new functional protein differently than Behe and Snoke in a couple of important ways, but they broadly point to the fact that there are other plausible routes than the restricted one(s) Behe and Snoke came up with - as I have alluded to before. Unlike them, Lynch does not assume that each individual mutation required for the evolution of a new function would be detrimental to the original function (which is why Behe and Snoke argue that a gene duplication is required before any mutations can take place towards new functionality). They can be neutral, hence the mutations could begin to accumulate (even by drift) before the duplication event. This difference already gives rise to 7 possible routes evolution could take for the evolution of a new functional protein by gene duplication (if 2 mutations are required to achieve a new function), see the figure below.“In a recent paper in this journal, Behe and Snoke (2004) questioned whether the evolution of protein functions dependent on multiple amino acid residues can be explained in terms of Darwinian processes. Although an alternative mechanism for protein evolution was not provided, the authors are leading proponents of the idea that some sort of external force, unknown to today's scientists, is necessary to explain the complexities of the natural world (Behe 1996; Snoke 2003). The following is a formal evaluation of their assertion that point-mutation processes are incapable of promoting the evolution of complex adaptations associated with protein sequences. It will be shown that the contrarian interpretations of Behe and Snoke are entirely an artifact of incorrect biological assumptions and unjustified mathematical oversimplification.”
The vertical lines represent the mutations that are needed to change the function. #2 is an example where the mutation happened before the duplication event. You might be thinking that Lynch is wrong to make the assumption of neutrality and that Behe and Snoke are right to assume that each of the mutations towards the new function would be detrimental to the old function. In fact, that’s exactly what Behe and Snoke claimed in their response to Lynch (which LSS also quoted in his video):
In this response paper, they make no references to support this statement. Lynch references multiple papers to justify this assumption, which also refute the one made by Behe and Snoke. He also points out that the papers that Behe and Snoke cited in their original paper actually don’t support their claim that the majority of aa substitutions decrease protein function.“Experimental studies contradict Lynch’s assumption of complete neutrality as a rule; the majority of amino acid substitutions decrease protein function.”
“Second, Behe and Snoke assume that all mutational changes contributing to the origin of a new multi-residue function must arise after the duplication process. They justify this assumption by stating that the majority of nonneutral point mutations to a gene yield a nonfunctional protein. To stretch this statement to imply that all amino acid changes lead to nonfunctionalization is a gross mischaracterization of one of the major conclusions from studies on protein biology—most protein-coding genes are tolerant of a broad spectrum of amino acid substitutions (Kimura 1983; Taverna and Goldstein 2002a,b). For example, in a large mutagenesis screen, Suckow et al. (1996) found that >44% of amino acid positions in the Lac repressor of Escherichia coli are tolerant of replacement substitutions. Axe et al. (1998) found that only 14% of amino acid sites in a bacterial ribonuclease are subject to inactivation by some replacement substitutions, with only one site being entirely nonsubstitutable. For human 3-methyladenine DNA glycosylase, ~66% of single amino acid substitutions retain function (Guo et al. 2004). Even for the highly conserved catalytic core regions of proteins, approximately one-third of amino acid sites can tolerate substitutions (Materon and Palzkill 2001; Guo et al. 2004). Many other studies (e.g., Kim et al. 1998; Akanuma et al. 2002), including all of those cited by Behe and Snoke, have obtained results of this nature. A deeper understanding of the fraction of amino-acid-altering mutations that have mild enough effects to permit persistence in a population comes from observations on within- and between-species variation in protein sequences (Li 1997; Keightley and Eyre-Walker 2000; Fay and Wu 2003), which generally indicate that 10% to 50% of replacement mutations are capable of being maintained within populations at moderate frequencies by selection-mutation balance and/or going to fixation. Because there is strong heterogeneity of substitution rates among amino acid sites (Yang 1996), these average constraint levels should not be generalized across all sites, many of which evolve at rates close to neutrality. Thus, most proteins in all organisms harbor tens to hundreds of amino acid sites available for evolutionary modification prior to gene duplication.”
As he pointed out, the functionality of proteins are often very tolerant to changes in individual amino acids, which is a form of “robustness”. This fact has been known for a long time, since the 1990s when James Well’s developed the “Alanine Scanning” technique. This involves systematically substituting each amino acid in a protein one-by-one with alanine (the most simplest amino acid), and observing how it affected the protein function. It generally showed that there usually are only a limited number of residues that are REALLY important to the function, while mutations at the other site are mostly tolerable. A more intensive “scanning” process is substituting each site systematically with all of the 20 amino acids (of course not at the same time). Here is an example of HIV-1 protease, shown in the figures below.
The image (a mutation sensitivity map) shows that most sites are fairly resistant against most aa substitutions, which is why the image on top shows mostly yellow colours, indicating the substitution didn’t have much effect. Only the ones that are rather darkly coloured represent the mutations that would have a strong negative effect on the protein function. So, Behe and Snoke, and thus LSS quoting them, are just wrong when they claim “the majority of amino acid substitutions decrease protein function”.
Lynch also includes another variable called “n”, which is the number of amino acids which could potentially contribute to a new function after a mutation. Behe and Snoke assumed that is only one combination possible that would lead to a new function, so if at minimum 2 mutations are needed then n=2, if there are 3 mutations needed, n=3. Here, Lynch modeled the numbers to be 10 or as high as 50 (which is significant as you will see next). Most of the other parameters were the same as Behe and Snoke (e.g. mutation rate of 10^-8). His results are summarised in this figure.
Lynch’s model shows that (when n=50) you would only need a population size of a little more than 1 million in order for a new functional protein to appear and be fixated within 1 million years on average. Contrast this to Behe and Snoke’s model, which would conclude that you need a population size of 1 trillion for the same fixation time of 1 million years, a difference of six orders of magnitude. There are more subtle differences between the models than I mentioned. You can see more problems that have been pointed out by other commentators in the editorial from the journal [or see Lauran Moran’s blog posts 1 and 2 below]. However, I covered the main differences in their assumptions which account for the majority of this discrepancy between the models of Lynch and Behe/Snoke. And, as previously shown, Lynch’s assumption of neutrality is better supported. It should also be stated that Lynch has stated that even his model is still quite restrictive, as there are other possible ways for new genes to evolve a new function following gene duplication that he hasn’t accounted for in his model.
"In summary, the conclusions derived from the current study are based on a model that is quite restrictive with respect to the requirements for the establishment of new protein functions, and this very likely has led to order-of-magnitude underestimates of the rate of origin of new gene functions following duplication. Yet, the probabilities of neofunctionalization reported here are already much greater than those suggested by Behe and Snoke. Thus, it is clear that conventional population-genetic principles embedded within a Darwinian framework of descent with modification are fully adequate to explain the origin of complex protein functions."
To further support Lynch’s assumptions, there was an laboratory experiment wherein they observed the process of gene duplication and the evolution of a new function that is (though not identical to, bus still) concordant with Lynch’s model. This was documented in the 2012 paper by Nasvall et al. Their observation centered around the gene HisA, which codes for the enzyme that catalyzes the production of histidine. From this gene, they have obtained a mutated version which is, in addition to its original function, is also capable of catalyzing the production of Tryptophan, but very poorly so. While this initial step was artificially induced by the scientists (which is undoubtedly what ID-proponents will point to dismiss this paper outright. The old “See, it had to happen in a lab. It was intelligent design after all!!”). However it shows that mutations which cause a new function to a protein aren’t necessarily detrimental to the original function. Again, supporting Lynch’s assumption of neutrality and that the mutations towards the innovation happened BEFORE duplication, which is starkly different from the model assumed by Behe and Snoke. What they did next is introduce this gene into a species of Salmonella that lacks the normal genes to synthesise histidine and tryptophan, and they grew it in a media without histidine and tryptophan. Hence, the bacteria is entirely reliant on this single gene to obtain both essential amino acids. They also placed this gene such that it was encouraged to be duplicated. Since the tryptophan activity of this gene is rather weak, gene duplication would actually be a beneficial mutation since it increases the production simply by increasing gene dosage (the fact that gene duplication itself can provide a selective advantage is also something that Behe and Snoke didn’t consider). From this point on, they let the bacteria grow for 3000 generations and the results are pretty amazing. What they saw is that in many lineages, the gene got indeed duplicated, but also many lineages experienced mutations that made one duplicated version a lot better at producing tryptophan, while the other was either better at producing only histidine or both. Not only that, similar results happened in multiple lineages independently, see figure below.
Here blow is a graph that shows the different evolutionary routes these bacteria made regarding how they improved their genes.
The Y-axis shows how “good” a gene is at producing histidine, and the X-axis shows how “good” it is at producing tryptophan. The 4 graphs show 4 different ways how duplicated genes diverged from one another in both activities (the numbers show how many times these routes were taken independently). Note that there are sometimes different mutations that would lead to roughly the same result. This is concordant with another assumption that Lynch made. That there are multiple combinations of mutations to reach similar results - there are multiple routes to reach similar destinations.
So, in short Lynch is right. Behe and Snoke are wrong.
Behe v.s. Durrett & Schmidt.
Next we have the Durrett & Schmidt paper from 2008. In LSS’ first video on whale evolution, he brought this paper up to argue that the waiting time for “even just two simple beneficial mutations” is too long to have happened within the ~10 million years it took for whales to evolve from being fully terrestrial to fully aquatic. The evolutionists don’t have enough time for even 2 beneficial mutations to have for whale evolution, let alone all the other ones that would also be needed!!
Here is where he went wrong. His first mistakes was saying that the numbers “a few million years” for fruit flies and “200 million years” for humans were about two beneficial mutations, while they were in fact about two NEUTRAL mutations (for fruit flies) and one neutral and one beneficial mutation (for humans). But the bigger problem here is that this paper isn’t about the waiting time for ANY two beneficial mutations. It was about two PRESPECIFIED mutations. In Jackson Wheat’s first rebuttal, he pointed this out, using the analogy of the difference between the odds of ANYONE winning the lottery and a SPECIFIC individual winning the lottery. If there is only one player, then yes, the odds between them are the same, but in a lottery there are a lot of players, so the odds that ONE of them will win is substantially greater than if you calculate the odds of a specific person winning. Which should remind you of the previous topic of Lynch, where it was noted there are many other ways for beneficial mutations to occur. There are even multiple mutations that have the same effect.
In LSS’ response video to Jackson, he made several mistakes regarding this paper. He claimed that the 2008 Durrett and Schmidt paper attempted to disprove the model presented in the 2004 Behe and Snoke paper (which was previously discussed). This just shows that LSS has not read the paper, nor the responses to Durrett and Schmidt written by Behe himself. If he did, he didn’t understand any of it. This wasn’t the goal of Durrett and Schmidt at all. They only mentioned the Behe and Snoke paper more as a side note, while only pointing out a problem they observed additional to those pointed out by Lynch.
That’s the only comment they made regarding that paper. Their main criticism of Behe was made clear earlier in the paper, where Durrett and Schmidt applied their calculations to critique an argument Behe made in his book “The Edge of Evolution” from 2007. In that book, Behe used the example of mutations that made Plasmodium (the parasite responsible for Malaria) resistant to a particular drug, called Chloroquine...mmh...why does that name sound familiar? Studies of the early 2000s showed that, for this resistance phenotype, two mutations in the transporter protein PfCRT were required (a single mutation doesn’t confer resistance, both have to be present). Behe refers to this one mutation pair as the “chloroquine complexity cluster” (CCC). Behe claims a single mutation has the probability of 1 in 10^10. So, for the CCC double mutation, it's that number squared, i.e. 1 in 10^20. A very low probability. Behe argues this is possible in Plasmodium since it has a huge population size and short reproductive cycle. For something like this to happen in humans, however, Behe says that it takes on average “hundred million times ten million years” or 1000 trillion years. Durrett and Schmidt showed that this figure is 5 million times larger than what they (initially) calculated, which was 216 million years. Later, they acknowledged a correction by Behe, which divides the number by 30, so Behe’s numbers are still over 150.000 times larger than those of Durrett and Schmidt. This is what LSS referred to as the “Serious flaw”. It’s a big difference, yes...but nevertheless, Behe's number is still way over the top."We are certainly not the first to have criticized Behe's work. Lynch (2005) has written a rebuttal to Beheand Snoke (2004), which is widely cited by proponents of intelligent design (see the Wikipedia entry on Michael Behe). Behe and Snoke (2004) consider evolutionary steps that require changes in two amino acids and argue that to become fixed in 108 generations would require a population size of 109. One obvious problem with their analysis is that they do their calculations for N = 1 individual, ignoring the population genetic effects that produce the factor of √u2. Lynch (2005) also raises other objections."
In LSS’ response video to Jackson, he wrongly claimed that Durrett and Schmidt narrowed their focus on two specific mutations, because, as he said:
They restricted their focus on two specific mutations, because they wanted to address a topic brought up by Behe (which LSS is apparently unaware of), which concerned the waiting time of two SPECIFIC beneficial mutations. LSS simply misapplied that to the waiting time for ANY two beneficial mutations, which is thus (as explained) very different. But what were the problems with Behe’s estimation that Durrett and Schmidt pointed out? The main difference is caused by the fact that Behe assumes the two mutations HAD to happen simultaneously, while Durrett and Schmidt allowed for the first mutation to happen before the second one occurs. They allow this since they assume the first mutation is neutral (or weakly deleterious), which means it escapes purifying selection and allows the mutation to spread throughout the population before the second mutation occurs. In Behe’s first reply, he states that the mutation is likely to be deleterious. However, in their reply to behe, Durrett and Schmidt note that if the first mutation is slightly deleterious; it would increase the waiting time by a factor of 2 or 3. This is still far from what Behe estimated, so even if the first mutation is slightly deleterious, it happening first and spreading through drift before the second mutation happens is a more plausible evolutionary route.“Almost all possible single or double or triple mutations amongst sequence space will be useless it would be silly to think that just any two mutations in general would be adaptive the vast majority are completely ineffective or degradative this is why they chose to model it so specifically”
Of course, Behe insists in his second reply that it is very likely deleterious.
“If the first mutation is indeed deleterious, then Durrett and Schmidt (2008) applied the wrong model to the chloroquine-resistance protein. In fact, if the parasite with the first mutation is only 10% as fit as the unmutated parasite, then the population-spreading effect they calculate for neutral mutations is pretty much eliminated, as their own model for deleterious mutations shows. What do the authors say in their response about this possibility? “We leave it to biologists to debate whether the first PfCRT mutation is that strongly deleterious.” In other words, they don’t know; it is outside their interest as mathematicians. (Again, I appreciate their candor in saying so.) Assuming that the first mutation is seriously deleterious, then their calculation is off by a factor of 10^4. In conjunction with the first mistake of 30-fold, their calculation so far is off by five-and-a-half orders of magnitude.”
LSS strangely highlights this paragraph from Behe, while stating that “the empirical data suggests that the intermediate mutations are not neutral or harmless”. He includes no references to show this empirical data. Neither does Behe here in this paragraph. He assumes (as it says right there) it is likely seriously deleterious to come to his conclusions. We already noted in the previous topic that proteins are often very resistant to aa substitutions, so the assumption of neutrality is actually very plausible. But let’s put a pin on this and return to this later.
The next paragraph LSS highlights is this:
Behe points out that, there is a chance that the second mutation will be ineffective in making the 9/10 transcriptions-factor-binding (TFB) site a complete 10/10, because of the possibility of other mutations between the first and second mutation that would make the 9/10 incomplete TFB into a (for example) 8/10 incomplete or 7/10. However, as Durrett and Schmidt already pointed out, this assumes that there is only one individual with the first mutation. As previously noted, they allowed for the first mutation to spread throughout the population before the second one occurred. What Behe describes here would only make one individual with the first mutation incapable of (or very unlike to) successfully completing the second mutation, but since there is a whole population of other individuals with the first mutation, which still have the 9/10 TFB poised to become 10/10, it has no effect on the waiting time.“...the effective mutation rate for transforming the string with nine matches out of ten to a string with ten matches out of ten will be only one tenth of the basic digit-mutation rate. If the string is a hundred long, the effective mutation rate will be one-hundredth the basic rate, and so on. (This is very similar to the problem of mutating a duplicate gene to a new selectable function before it suffers a degradative mutation, which has been investigated by Lynch and co-workers.”
In Behe’s second response, he simply doesn’t accept the correction. He objects by stating that he wrote “no such thing” in his letter about “one individual”. Even if he didn’t explicitly say that, his objection would only count if there is only one individual with the initial mutation. Behe is simply stuck in his mindset that the two mutations had to have happened in the same individual at the same time. He thinks it just has to, since the first mutation is likely deleterious, but we covered that already.“In Behe (2009), the accompanying Letter to the Editors in this issue, Michael Behe writes (here and in what follows italicized quotes are from his letter), “… their model is incomplete on its own terms because it does not take into account the probability of one of the nine matching nucleotides in the region that is envisioned to become the new transcription-factor-binding site mutating to an incorrect nucleotide before the 10th mismatched codon mutates to the correct one.” This conclusion is simply wrong since it assumes that there is only one individual in the population with the first mutation. There are on the order of 1/u2^½ individuals with the first mutation before the second one occurs, and since this event removes only one individual from the group with the first mutation, it has no effect on the waiting time.”
The second to last last thing LSS highlights from Behe’s second reply this:
After highlighting this, LSS says that this “inappropriate multiplication of probabilistic resources” was a factor that made Durrett and Schmidts calculations off by seven orders of magnitude (highlighting a section from further back Behe’s reply). This is not true. This “multiplication of probabilistic resources” were NOT included in Durrett and Schmidts calculations at all. What happened was this. Behe observed that even according to their calculations, the waiting time for a pre-specified pair of mutations in humans is 216 million years. So, even taking Durrett and Schmidt’s calculations for granted, Behe noted that their calculations showed that still gives a prohibitively long waiting time.“The final conceptual error that Durrett and Schmidt commit is the gratuitous multiplication of probabilistic resources.”
However, as Durrett and Schmidt noted in their reply:“Durrett and Schmidt (2008, p. 1507) retort that my number “is 5 million times larger than the calculation we have just given” using their model (which nonetheless gives a prohibitively long waiting time of 216 million years).”
“Finally, Behe notes that for one prespecified pair of mutations in one gene in humans with the first one neutral, we obtain a “prohibitively long waiting time” of 216 million years. However, there are at least 20,000 genes in the human genome and for each gene tens if not hundreds of pairs of mutations that can occur in each one. Our results show that the waiting time for one pair of mutations is well approximated by an exponential distribution. If there are k nonoverlapping possibilities for double mutations, then by an elementary result in probability, the waiting time for the first occurrence is the minimum of k independent exponentials and hence has an exponential distribution with a mean that is divided by k. From this we see that, in the case in which the first mutant is neutral or mildy deleterious, double mutations can easily have caused a large number of changes in the human genome since our divergence from chimpanzees. Of course, if the first mutant already confers an advantage, then such changes are easier.
They pointed out the same thing that I previously did, several times. It’s the same mistake LSS originally made in his video about the Durrett & Schmidt paper. There are multiple possible mutation pairs that could be beneficial, besides one prespecified pair of mutations. This is what Behe refers to as “multiplication of probabilistic recourse”, which - again - was not included in their calculations, since - again - the aim was to calculate the waiting time for one pair of PRESPECIFIED beneficial mutations. Not ANY pair of beneficial mutations. Of course, Behe refused to accept this.
Which is exactly word-for-word what LSS said in response to Jackson. In the video, LSS doesn’t make it clear that he was directly quoting Behe, so LSS’ response was basically a copy paste from Behe. Regardless, the mistake being made here is that “almost all possible mutations will be useless” is taken to be equivalent to “there are only 2 possible mutations that would be beneficial”. Of course, I agree with the first one. The vast majority of possible mutations are “useless”. Let’s say that out of the billion possible mutations, 99.99% are either neutral or deleterious. Only 0.001% are beneficial. That would still mean 100.000 possible mutations are beneficial, far from being just two. Even if we focus one one specific phenotype, like Chloroquine There is more to say on this, but let’s put a pin on this as well for the finale.“The answer of course is that in almost any particular situation, almost all possible double mutations (and single mutations and triple mutations and so on) will be useless. Consider the chloroquine-resistance mutation in malaria. There are about 10^6 possible single amino acid mutations in malarial parasite proteins, and 10^12 possible double amino acid mutations (where the changes could be in any two proteins). Yet only a handful are known to be useful to the parasite in fending off the antibiotic, and only one is very effective — the multiple changes in PfCRT. It would be silly to think that just any two mutations would help. The vast majority are completely ineffective. Nonetheless, it is a common conceptual mistake to naively multiply postulated “helpful mutations” when the numbers initially show too few.”
One last thing I want to cover from LSS is this statement:
That last bit is where LSS cites the 2004 paper by White on antimalarial drug resistance.This is where Behe gets his 1 in 10^20 figure from for odds of the double mutation to occur. However, the big mistake Behe makes is that this isn’t actually the odds for the double mutation to occur that causes the pathogen to be resistant to Chloroquine. It is the probability of the mutation occurring TIMES the probability of it spreading throughout the population enough to reach a frequency that will be detected. The actual mutation rate per site in the species Plasmodium falciparum is 2.5x10^-9, so for the double mutation to happen it is (that number squared =) 6.25x10^18. Although, the typical mutation rate per site for eukaryotes is about 1 10^10, so the 1 in 10^20 for a SPECIFIC double mutation is mostly correct. But it is important to note that Behe doesn’t recognise the difference between the odds of just the double mutation occurring and the odds of that in addition to the odds of it increasing in frequency within a population to the point when it is detected.“When correcting for these errors, and others, their model agrees quite well with the original paper by Behe and Snoke, as well as the empirical public health data and the literature.”
Now we get to the finale! I have pinned two points for this end paragraph, which are the most important points of contention between Behe and Durrett & Schmidt.
- Whether the first mutation of the double mutation, that is required for the beneficial phenotype of Chloroquine resistance in the Malaria pathogen, is neutral (so they don’t have to occur at the same time) or strongly deleterious (so they do have to occur at the same time).
- Whether there are more than one pair of possible mutations that could lead to the same phenotype, i.e. whether there are multiple ways for X to evolve.
It shows different strains that have different variants of the PfCRT gene, which have different levels of Chloroquine (CQ) uptake capabilities (i.e. how high they are positioned on the figure). The differences between these variants are single aa substitutions, which are shown as codes (i.e +75E). The arrows basically show how chloroquine resistance evolved via a series of strains, each acquiring one specific mutation at a time. The coloured arrows indicate whether the mutation was beneficial (green), detrimental (red) or neutral (blue). Note that many mutations here are neutral, even the ones occurring after the starting point on the bottom left (HB3). Hence, Behe was wrong when he claimed the first mutation is deleterious.
CORRECTION: I have to make a correction after I saw Ration alMind's comments (somewhere else, not here on LoR). I mistakingly took the CQ uptake capacity to be equal to fitness. The coloured lines in the figure denote whether the mutation is neutral/beneficial/detrimental with respect to Chloroquine uptake, but that doesn't mean overall fitness. The paper actually references studies that shows that strains, possessing the mutations for increase CQ uptake, are actually worse-off than the wild-type in the absence of CQ. So there seems to be a trade-off between CQ uptake and the "normal" function of the PfCRT protein (similar to the one described in the BONUS paper).
More specifically, one of the mutations called K76T [meaning: Lysine at residue 76 replaced by Threonine] makes the malaria parasite "less viable" in the absence of other mutations, however the other mutations (without K76T) are not deleterious."Although all the routes originating from D17 (N75E-K76T-A220S-Q27IE) entail one or more transient decreases in the ability of PfCRT to transport CQ, some of the intermediate haplotypes with reduced CQ transport activities may in fact represent favorable tradeoffs between conferring a moderate level of CQ resistance and maintaining the normal physiological role of the protein, which is essential but as yet unknown (16, 20, 21). Indeed, PfCRT haplotypes of the ET lineage, particularly those containing M74I-N75E-K76T, are associated with a significant decrease in the fitness of the parasite, to the extent that these CQR strains are out-competed by wild-type CQS parasites when CQ is withdrawn (22, 23). Hence, it is possible that one or more of the intermediate haplotypes of the ET lineage possesses sufficient CQ transport activity to ensure survival of the parasite when exposed to medium levels of CQ pressure and that the additional mutations found in the GB4, K1, and Dd2 haplotypes further impair the normal function of the protein and are advantageous only when CQ use is high. Furthermore, it is important to note that mutations in PfCRT are likely to have been accompanied at various points by changes in other genes, some of which may have served to maintain or even increase the parasite’s resistance to CQ when a new mutation in PfCRT decreased its CQ transport activity. One such possible modulator is the multidrug resistance transporter 1 (PfMDR1), certain mutations in which have been shown to increase the in vitro CQ resistance of some (but not all) parasites harboring mutant PfCRT (24, 25). Other proteins that have been implicated in the CQ resistance phenotype and which also may have served to offset decreases in the CQ transport activity of PfCRT include the multidrug resistance-associated proteins 1 and 2 (PfMRP1 and PfMRP2) (26, 27)."
So this mutation could occur as the last mutation and confer a NET increase in fitness in the presence of the anti-malarial drug."These results suggest reduced parasite viability resulting from K76T in the absence of other pfcrt mutations. This situation is not reciprocal however, in that parasites harboring all the other mutations except for K76T (illustrated by our back-mutants) show no signs of reduced viability in culture."
Furthermore, a paper from 2016 shows how resistance can be achieved by multiple evolutionary routes of 4 mutations, which continuously increased fitness after each mutation. So even a single mutation could confer a slight increase in resistance (although at first very slightly).
The paper also emphasised how important it is for investigating the evolvability of particular phenotypes by modelling different selection pressures, not just looking at the effects of one mutation under a few environments.
There is even a wild variant 106/1, which possesses some of the mutations needed so it isn’t resistant yet, but it is able to become resistant by just one more mutation. Hence, Behe was also wrong when he claimed that the two mutations had to occur at the same time. Not only that, if you look closely at the two possible ways for HB3 to become D32, one path shows the mutations happening with first +75E then 76T, and the other shows first 76T then 76E. So not only don’t they have to be simultaneous, they can also happen in any order, something that even Durrett and Schmidt didn’t account for. You can also see that, since there isn’t just a single series of arrows, there are many ways for evolution to improve CQ uptake. Hence, Behe was also wrong when he only allowed evolution to take one specific way to reach the destination of chloroquine resistance.
It is funny to note that this figure shows the step-by-step evolution of something, which Behe considers too improbable to have evolved. How Behe, the champion of irreducible complexity, can look at this paper and say “yeah, that supports my position” is just beyond me.
This should be also a lesson for many people who accept evolution out there. Natural selection is important, certainly, but it isn’t the only significant aspect of it, not even when it comes to adaptation. As you can see here, some of the necessary intermediate steps are neutral, or sometimes even deleterious. These mutations that are required, but themselves not sufficient, are often dubbed “potentiating” mutations. Selection obviously wouldn’t promote these, but drift makes it possible for some to spread and remain in a population until further mutation occurs that “completes” the phenotype. So not every incremental step HAS to be beneficial for evolution, they can be neutral (or even deleterious). The second lesson is that when we observe, in hindsight, the many mutations that were involved in producing a beneficial trait, that doesn’t mean that these are the ONLY mutations that could have produced a beneficial trait (not even the same beneficial trait) or that they HAD to occur in one particular order. As we have seen, even for the same phenotype (e.g. CQ resistance), there are multiple routes evolution could take and could've taken. I previously described this fallacy with the lottery analogy. This is a common cognitive pitfall that, when people note in hindsight an extremely unlikely event, it makes them think it couldn’t have happened by chance alone. In foresight, most of all extremely unlikely events that you could prescribe will never occur, but since there are so many of these, extremely unlikely events occuring is a mathematical certainty.
EDIT: And a third lesson (also for myself, regarding the correction), whether a mutation is neutral, beneficial or deleterious is often dependent on the environment. Under environmental conditions A, B, C they may be beneficial, but under X, Y and Z, they may be deleterious.
So when someone (creationist or evolutionist) comes to you and says that EVERY incremental step had to infer some advantage, or if they say that X evolved via route Y, route Y was the ONLY way for X to have evolved, please kindly introduce them to this thread for me.
So that’s it. My apologies for the long post. I will leave you all with the citations below, plus a bonus paper (from 2019) that I found while doing research on this. It is on the evolution of drug resistance in Malaria and also on the same PfCRT gene, but here it shows that mutations to this gene can make it resistant to different types of drugs, but not “optimally” for both, i.e. mutations that make it better resistant to one drug makes it less resistance to the other drug as well. A great example of evolutionary trade-offs/constraints. It’s a good read.
CITATIONS
Citations for Behe v.s. Lynch
Main
2004 Behe and Snoke: Simulating evolution by gene duplication of protein features that require multiple amino acid residues
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2286568/
2005 Lynch: Simple evolutionary pathways to complex proteins
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253472/
2005 Behe: A response to Michael Lynch
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253464/
2005 Editorial and position papers
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2253483/
Further
1989 High-Resolution Epitope Mapping of hGH-Receptor Interactions by Alanine-Scanning Mutagenesis
https://sci-hub.tw/10.1126/science.2471267
2018 Mutation Sensitivity Maps: Identifying Residue Substitutions That Impact Protein Structure Via a Rigidity Analysis In Silico Mutation Approach
https://www.liebertpub.com/doi/10.1089/cmb.2017.0165?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub 0pubmed
2004 White: Animal drug resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC385418/
Extra: Blog posts by Laurence Moran, which explains the subject more briefly and is easier to understand than the technical literature above.
1. Waiting for multiple mutations: Intelligent Design Creationism v. population genetics
[Explains the Behe and Snoke paper]
https://sandwalk.blogspot.com/2015/12/waiting-for-multiple-mutations.html
2. Waiting for multiple mutations: Michael Lynch v. Michael Behe
[Explains Lynch’s response paper to Behe and Snoke, and also briefly mentions Durrett and Schmidt]
https://sandwalk.blogspot.com/2015/12/waiting-for-multiple-mutations-michael.html
Citations for Behe v.s. Durrett & Schmidt
2008 Durrett and Schmidt: Waiting for Two Mutations: With Applications to Regulatory Sequence Evolution and the Limits of Darwinian Evolution
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2581952/
2009 Behe: Waiting longer for two mutations
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2644969/
2009 Durrett and Schmidt: Reply to Behe
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2644970/
2009 Behe: A roll-up of "Waiting Longer for Two Mutations", Parts 1-5
https://www.discovery.org/a/9611/
2005 A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1173140/
2014 Summers: Diverse mutational pathways converge on saturable chloroquine transport via the malaria parasite’s chloroquine resistance transporter
https://doi.org/10.1073/pnas.1322965111
2016 Combinatorial Genetic Modeling of pfcrt-Mediated Drug Resistance Evolution in Plasmodium falciparum
https://doi.org/10.1093/molbev/msw037
Extra: Blog posts by Laurence Moran, PZ Myers and Kenneth Miller
2012 Moran: Understanding Mutation Rates and Evolution
https://sandwalk.blogspot.com/2012/01/understanding-mutation-rates-and.html
2014 Moran: Taking the Behe challenge!
https://sandwalk.blogspot.com/2014/08/taking-behe-challenge.html
2014 Moran: Flunking the Behe challenge!
https://sandwalk.blogspot.com/2014/08/flunkiung-behe-challenge.html
2014 Moran: CCC's and the edge of evolution
https://sandwalk.blogspot.com/2014/08/cccs-and-edge-of-evolution.html
2014 Moran: Michael Behe's final thoughts on the edge of evolution
https://sandwalk.blogspot.com/2014/08/michael-behes-final-thoughts-on-edge-of.html
2014 Moran: Understanding Michael Behe
https://sandwalk.blogspot.com/2014/08/understanding-michael-behe.html
2014 Moran: On the irrelevance of Michael Behe
https://sandwalk.blogspot.com/2014/12/on-irrelevance-of-michael-behe.html
2016 Moran: Revisiting Michael Behe's challenge and revealing a closed mind
https://sandwalk.blogspot.com/2016/12/revisiting-michael-behes-challenge-and.html
2016 Moran: Targets, arrows, and the lottery fallacy
https://sandwalk.blogspot.com/2016/01/targets-arrows-and-lottery-fallacy.html
2014 PZ Myers: Quote-mined by Casey Luskin!
https://freethoughtblogs.com/pharyngula/2014/07/17/quote-mined-by-casey-luskin/
2014 PZ Myers: Aren’t we all more than a little tired of Michael Behe?
https://freethoughtblogs.com/pharyn...all-more-than-a-little-tired-of-michael-behe/
2007 Kenneth Miller: Falling over the edge
https://www.nature.com/articles/4471055a
2014 Kenneth Miller: Edging towards irrelevance
http://www.millerandlevine.com/evolution/behe-2014/Behe-1.html
BONUS PAPER
2019 Nature News “Malaria parasites fine-tune mutations to resist drugs”
https://www.nature.com/articles/d41586-019-03587-0
2019 Paper: Structure and drug resistance of the Plasmodium falciparum transporter PfCRT
https://doi.org/10.1038/s41586-019-1795-x
Last edited: